, Nitin J Peters1
, Shailesh Solanki1
, Amita Trehan2
, Kirti Gupta3
, Monika Bawa1
, Nitin J Peters1
, Shailesh Solanki1
, Amita Trehan2
, Kirti Gupta3
, Monika Bawa1

Hirschsprung disease (HSCR) is a genetic disorder with an incidence of 1:5000, seen in the pediatric age group. The association between HSCR and neuroblastoma (NBL), ends of the neurocristopathy spectrum is rare. Less than 10 cases of this association are reported in the literature and the association between the Phox gene and Sox10 gene in the pathophysiology of these is being studied. We report a one-year-old baby, who presented to us, with chronic constipation on regular enemas and laxative usage. There was a history of delayed passage of meconium. At the time of Duhamel’s pull through a well-defined, bilobed hard presacral mass, was encountered. Excision and coccygectomy were done and the pull was completed. The histopathology showed a well-differentiated NBL. Fludeoxyglucose positron emission tomography scan and the N-Myc amplification were negative and the patient was managed with expectant treatment. She is doing well over a 3-year follow-up with no recurrence and good resolution of bowel functions.
Hirschsprung disease (HSCR) is a genetic disorder with an incidence of 1:5000, seen in the pediatric age group. The association between HSCR and neuroblastoma (NBL), ends of the neurocristopathy spectrum is rare. Less than 10 cases of this association are reported in the literature and the association between the Phox gene and Sox10 gene in the pathophysiology of these is being studied. We report a one-year-old baby, who presented to us, with chronic constipation on regular enemas and laxative usage. There was a history of delayed passage of meconium. At the time of Duhamel’s pull through a well-defined, bilobed hard presacral mass, was encountered. Excision and coccygectomy were done and the pull was completed. The histopathology showed a well-differentiated NBL. Fludeoxyglucose positron emission tomography scan and the N-Myc amplification were negative and the patient was managed with expectant treatment. She is doing well over a 3-year follow-up with no recurrence and good resolution of bowel functions.
Hirschsprung disease (HSCR) is a common genetic disorder in the pediatric age group with an incidence of 1:5,000 [1]. It falls under the spectrum of neurocristopathies (NCP), which are a group of disorders that occur due to the failure of neural crest cells (NCC) to migrate, proliferate, or differentiate during embryogenesis [2]. Neurocristopathy can affect multiple organ systems and present in various ways. The commonly seen neurocristopathies are HSCR, neuroblastoma (NBL), and central hypoventilation syndrome. To the best of our knowledge only seven cases of HSCR associated with NBL, have been reported in English literature (Table 1) [3, 4, 5, 6, 7, 8].
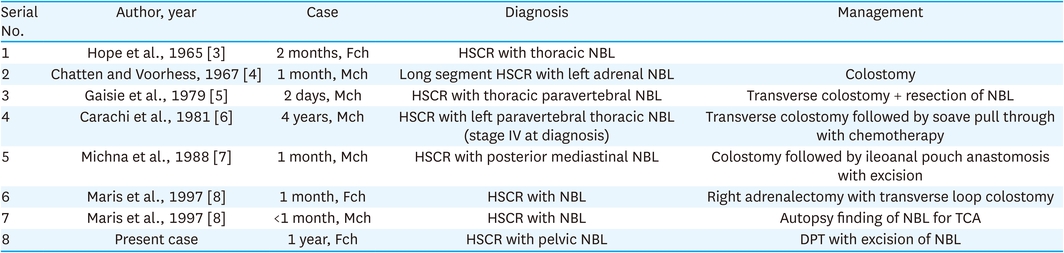
Table 1
Table showing the reported cases of HSCR with NBL
A one-year-old baby presented with chronic constipation and abdominal distension since birth. The child was a full-term baby with a history of delayed passage of meconium. The child also had infrequent episodes of urinary retention. Clinical examination showed a distended abdomen; on the per rectal examination, the rectum was loaded with faeces. Routine laboratory tests and thyroid function tests were normal. On abdomen X-rays, dilated large bowel loops were seen; retrospectively, no mass/opacification was seen on the X-rays. Urological investigations like renal ultrasound and micturating cystourethrogram were normal (Fig. 1).
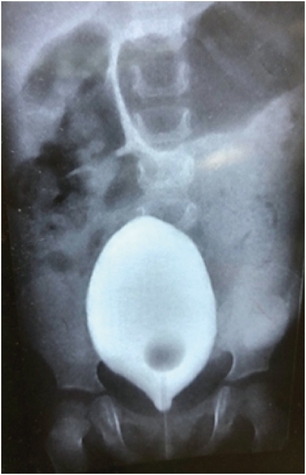
Fig. 1
Showing the micturating cystourethrography (urinary bladder) good capacity, uniform, no reflux, and dilated bowel loops can be noted.
In view of the grossly distended abdomen, the routine contrast enema was not done. But suspecting it to be an HSCR, a divided sigmoid colostomy with a rectal biopsy was done as per our institutional protocol [9]. The biopsy report confirmed HSCR as it showed absent ganglion cells in the rectum. The child was nutritionally built up and after adequate weight gain, was taken up for a definitive pull-through. Modified Duhamel’s pull-through was carried out as the proximal stoma was ganglionated as per the HPE report. During the retro-rectal tunnelling, a firm bilobed presacral mass of size 6×8 cm was encountered (Fig. 2A and B). Complete excision with coccygectomy was done simultaneously assuming the mass to be a presacral teratoma.
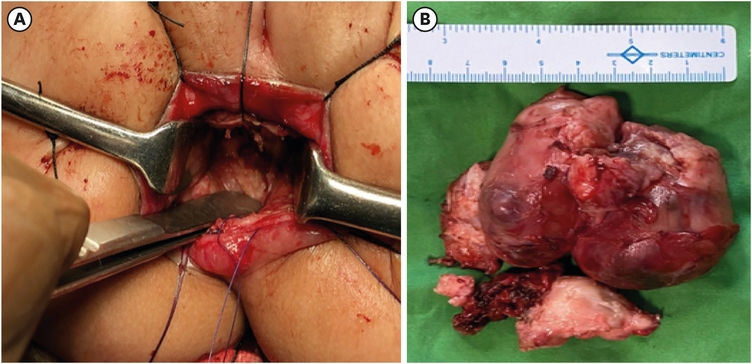
Fig. 2
Intraoperative images. (A) Showing the presacral mass which is 6×8 cm bilobed, firm, irregular in outline. (B) Image shows the excised specimen of the neuroblastoma.
Post-operatively, the child was orally allowed on postoperative day 4 and was passing stools adequately. The histopathology report of the presacral mass showed a well-differentiated NBL, negative for N-Myc amplification. Fludeoxyglucose positron emission tomography scan done post-surgery was suggestive of a faint somatostatin receptor-expressing lesion in the sacrococcygeal area and pre-coccygeal lymph nodes. The NBL fell into the low-risk category and is under close follow-up with ultrasonography quarterly in the first year and semiannually in the second year. She is doing well over a 3-year follow-up with no recurrence and good resolution of bowel and bladder functions.
The study was performed according to the Helsinki guidelines and consent for publication of the report and associated images has been taken and signed by the guardian.
We report a rare association of HSCR and NBL, which fall at the extremes of the spectrum of neurocristopathy. We attribute it to the genetics and embryology of NCCs rather than a chance association between them.
HSCR is distinguished by the lack of enteric ganglion cells in the large intestine. The most common site is the recto-sigmoid junction, but it can occur at various lengths of the large intestine. The etiology of HSCR is multi-factorial and genetics play a predominant role. The NCCs fail to migrate in HSCR and lead to a lack of ganglion cells in the large intestine. Mutations in the signalling pathway of the RET proto-oncogene play a pivotal role in the development of HSCR [10]. RET mutation on chromosome 10q11.2 is seen in 50% and 20%–30% of familial HSCR and sporadic HSCR cases respectively [1]. Isolated HSCR is most common with an incidence of 70%. Syndromic or chromosomal anomalies, trisomy 21 being the most common, are seen in 30% of cases of HSCR.
NBL is the most common extracranial malignant tumour and accounts for 15% of overall mortality due to malignancy in children. The median age of presentation is 18 months. NBL arises within the neuronal ganglia of the peripheral sympathetic nervous system, which are derived from the ventrolateral NCCs. Abnormal differentiation of NCCs in the adrenal medulla and sympathetic chain ganglia results in NBL [11].
NCCs undergo an “epithelial to mesenchymal transition” into a surfeit of distinct tissues, like the sympathoadrenal lineages. Gaisie et al. [5] proposed that during the process of embryogenesis, NCCs multiply and migrate to different parts of the body and differentiate into various sensory ganglia like sympathetic, parasympathetic, enteric ganglia, chromaffin system, adrenal gland, etc. Interruption of this normal developmental process due to mutations of RET oncogene causes HSCR, while interruption due to mutations in MYCN or ALK oncogenes, causes NBL [12].
Approximately, 1.5% and 10% of patients with HSCR and total colonic aganglionosis (TCA) respectively, have an association with congenital central hypoventilation syndrome (CCHS). However, 50% of CCHS patients will have HSCR, and this association is known as Haddad syndrome. HSCR associated with CCHS usually have long segment involvement and TCA is nearly 60%. NBL is seen in 20% of patients with Haddad syndrome [13]. Clausen et al. [14], in 1989 reported a family with ganglioneuromas in a lady, NBLs in her 2 daughters, coexisting with von Recklinghausen’s neurofibromatosis, HSCR, and the jaw-winking syndrome in another family member, which further corroborates the genetic and chromosomal association of these diseases.
The ongoing research has proved heterozygous “non-Polyalanine repeat mutations (NPARM) of the paired-like homeobox 2b (Phox2b) gene” as seen in HSCR and NBL (Fig. 3). The proliferation and differentiation of NCCs are monitored by the expression of Phox2b and sex-determining region Y box10 (Sox10). Both Phox2b and Sox10 expression is observed on undifferentiated NCCs. However, cells with neuronal differentiation express only Phox2b and those with glial cell differentiation express Sox10 alone. Lower Sox10 levels induce Phox2b thereby maintaining the neurogenic potential, while higher Sox10 levels promote gliogenesis and inhibit neuronal differentiation. Dysregulation of Sox10 by NPARM Phox2b can result in autonomic neurocristopathy. The defects in Phox2b occur in 15%–20% of HSCR cases and some cases of NBL [15]. In our index case, genetic studies have not been done, but the co-existence may not merely be a coincidence but maybe a variant of neurocristopathies.
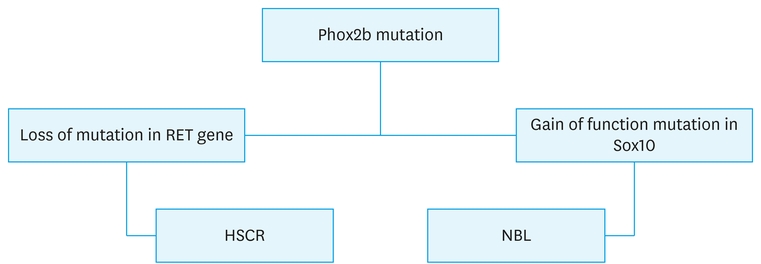
Fig. 3
Algorithm of Phox2b mutation causing HSCR and NBL.
HSCR, Hirschsprung disease; NBL, neuroblastoma.
The management and outcome in patients with neurocristopathies may be different from their isolated counterparts. Nemecek et al. [11] observed that NBL associated with other neurocristopathies were multifocal, metastatic, unfavourable histology, and poor long-term survival. The patients died because of the progression of the disease or respiratory failure, especially those associated with CCHS [11]. TCA and total ileal aganglionosis, are more common in neurocristopathies, with an incidence of 59%, and may lead to difficult management and an increase in morbidity and mortality [16].
In conclusion, NBL in association with HSCR is extremely rare. The association may be attributed to the common embryonic origin from neural crest cells. The Phox2b and Sox10 pathways may hold more answers to this uncommon association.
Conflict of Interest:No potential conflict of interest relevant to this article was reported.
Author Contributions:
Conceptualization: V.S., P.N.J.
Data curation: V.S., P.N.J.
Formal analysis: V.S., P.N.J.
Supervision: P.N.J., S.S., T.A., G.K., B.M.
Validation: P.N.J., S.S., B.M.
Visualization: S.S., B.M.
Writing - original draft: V.S., P.N.J.
Writing - review & editing: V.S., P.N.J., S.S., T.A., B.M.
The authors thank the patient and her parents for their support and patience in helping us with our research efforts. We are indebted to many members of the staff for taking care of the patient. We thank our colleagues in hemato-oncology, radiology, and pathology who participated in the management of this patient.