ABSTRACT
The concurrent occurrence of colonic atresia, malrotation, and Hirschsprung’s disease in neonates is extremely rare. These anomalies often share embryologic origins and present overlapping clinical symptoms that complicate diagnosis and management. We report two neonatal cases with this rare triad. Case 1 involved a term neonate initially diagnosed with esophageal atresia and later found to have colonic atresia, malrotation, and Hirschsprung’s disease. Case 2 was a preterm neonate presenting with abdominal distension and perforation, ultimately diagnosed with the same triad. Both underwent staged surgical management, including Duhamel’s procedures after confirming aganglionosis. Awareness of the possible coexistence of these anomalies is essential in neonates with colonic atresia and non-fixed colon. Surgical planning should anticipate aganglionosis and include rectal biopsy. This report emphasizes the importance of early suspicion and multidisciplinary approach for optimal outcomes.
-
Keywords: Colonic atresia; Hirschsprung disease; Intestinal malrotation; Neonate
INTRODUCTION
The coexistence of intestinal atresia, malrotation, and Hirschsprung’s disease represents a rare but insightful example of how disruptions during embryonic development can manifest in interconnected anomalies. Each of these conditions arises during overlapping developmental windows, making them susceptible to shared etiological factors such as vascular compromise, genetic mutations, or environmental insults [
1-
3]. We report the cases of two newborns presenting with colonic atresia, malrotation, and Hirschsprung’s disease in coexistence.
CASE REPORT
1. Case 1
A 2,710 g female neonate was born at 38+4 weeks of gestation. Prenatal ultrasound findings included suspicion of meconium peritonitis, left ectopic kidney, and cardiac anomalies. Postnatally, the abdomen was soft and non-distended. However, the failure to pass a gavage tube beyond 10 cm raised suspicion of esophageal atresia and chest radiography confirmed the diagnosis. Abdominal ultrasonography revealed distended pelvic bowel loops and a collapsed rectum without signs of meconium peritonitis. The neonate was admitted to the neonatal intensive care unit (NICU), remained nil per os, and showed no meconium passage.
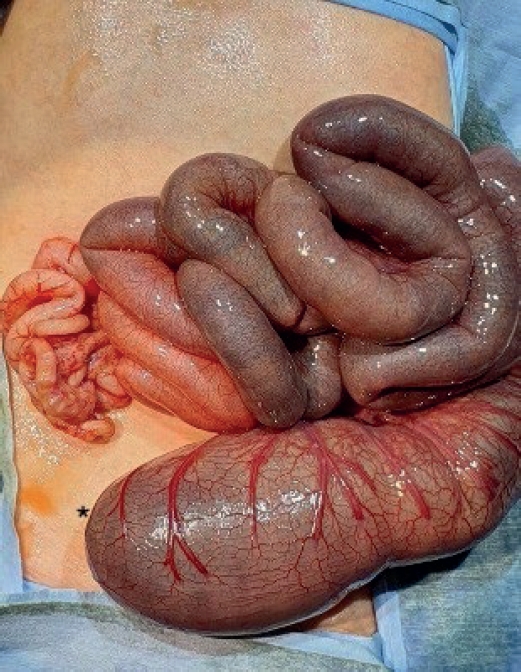
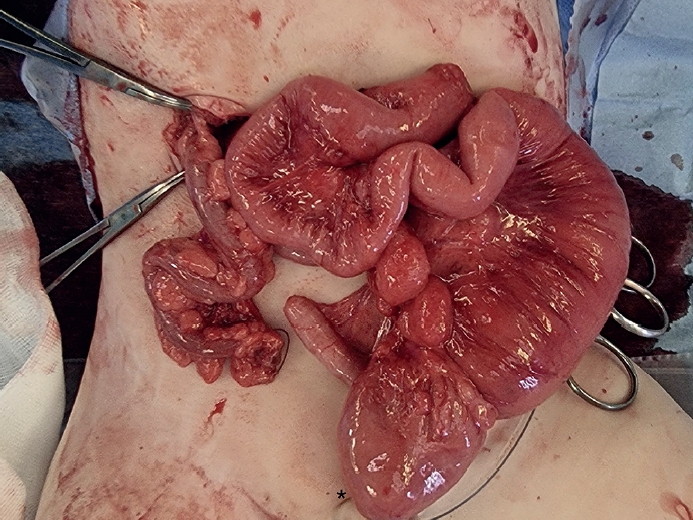
On day 3, thoracoscopic tracheoesophageal fistula ligation and esophago-esophagostomy were performed uneventfully. On postoperative day (POD) 2, the infant progressively developed abdominal distension. Imaging revealed dilated bowel loops, and an emergency laparotomy was conducted. Surgical findings included distal ileal atresia and colonic atresia with absence of the cecum and ileocecal valve, along with non-rotated, non-fixated colon distal to the atresia (
Fig. 1). End-to-end ileocolostomy was performed.
Barium esophagography on POD 10 confirmed an intact esophageal repair, and oral feeding was initiated. Despite passing stool daily, the infant demonstrated persistent feeding intolerance and persistent abdominal distension. Colon studies on PODs 22 and 50 identified contrast passage at the anastomosis with an unused microcolon. A second laparotomy on POD 55 uncovered abrupt narrowing around the ileocolonic anastomosis, prompting resection of a short bowel segment and creation of a double-barrel ileostomy. Biopsy of the resected bowel demonstrated the absence of ganglion cells in the colonic side, while the ileal segment exhibited ganglion cells. Rectal full thickness biopsy confirmed Hirschsprung’s disease.
The neonate tolerated full oral feeding postoperatively and was discharged in stable condition. At 7 months, laparoscopic total colectomy and ileal resection with rectorectal pull-through anastomosis (Duhamel’s procedure) was performed. The patient showed good stool passage postoperatively, achieved full enteral feeding, and was discharged in stable condition.
2. Case 2
A 1,920 g male neonate was born at 35+0 weeks of gestation. There were no abnormal findings on the prenatal ultrasound. Postnatally, the neonate had a distended abdomen, greenish vomiting, and did not pass meconium. Abdominal ultrasonography revealed distended bowel loops with a collapsed gasless rectum and the neonate was admitted to the NICU.
On day 3, the infant progressively developed abdominal distension. Peritoneal free air was detected on abdominal X-ray, and emergency laparotomy was performed. Surgical finding included colonic atresia with mesenteric defect at proximal ascending colon with small bowel perforation at 20 cm above the ileocecal valve. A transition zone was seen 30 cm above the ileocecal valve and loop ileostomy was performed at this site. Biopsy of the ileostomy bowel exhibited ganglion cells while the colon distal to the atresia showed absence of ganglion cells.
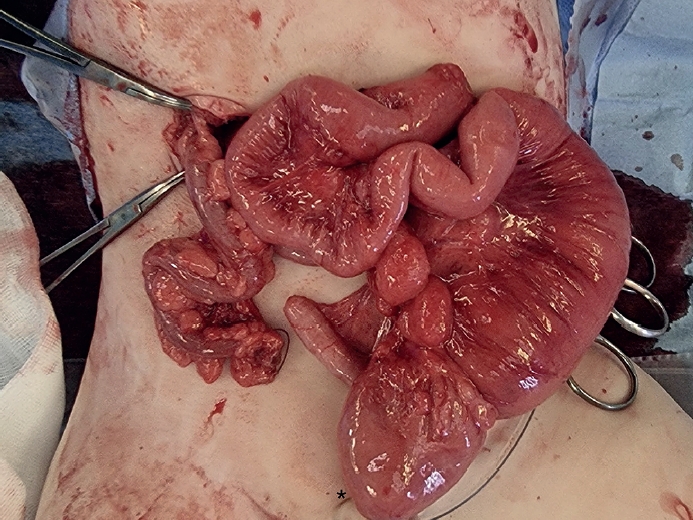
At 10 months, laparoscopic total colectomy and ileal resection with rectorectal pull-through anastomosis (Duhamel’s procedure) was performed. Surgical findings included colonic atresia with complete malrotation and non-rotated, non-fixated colon distal to the atresia (
Fig. 2). Frozen section biopsy was performed during the surgical procedure and showed that there was ganglion cells present in the bowel segment (terminal ileum, proximal ascending colon) between the ileostomy and the colonic atresia. No ganglion cells were seen in the distal colon stump and rectum, which confirmed the diagnosis of Hirschsprung’s disease. Intraoperative findings confirming colonic atresia with malrotation and distal aganglionosis are shown in
Fig. 2.
Postoperatively, the patient passed stool well and successfully achieved full enteral feeding. On POD 14, the infant was discharged in stable condition.
DISCUSSION
During the 5th to 12th weeks of gestation, several critical processes occur in the gastrointestinal system. Neural crest cells migrate cranio-caudally to populate the gut wall with ganglion cells, forming the enteric nervous system [
4,
5]. This process is essential for coordinated peristalsis and gastrointestinal function. Simultaneously, the midgut elongates, herniates into the umbilical cord, and undergoes a 270° counterclockwise rotation around the superior mesenteric artery to achieve proper orientation [
2,
3]. By the 10th to 12th weeks, the gut returns to the abdominal cavity, where retroperitoneal fixation and final neuroenteric patterning occur. Disruptions during these stages, especially vascular insults or ischemia, can lead to profound developmental abnormalities. For instance, ischemic events may halt bowel development, causing atresia, or block neural crest migration, resulting in aganglionosis seen in Hirschsprung’s disease [
1,
6]. Similarly, insufficient vascular support or mesenteric abnormalities can interfere with midgut rotation, leading to malrotation. These anomalies are not coincidental; they may share common pathogenetic mechanisms. Colonic atresia can obstruct the caudal migration of neural crest cells, exacerbating aganglionosis, while intestinal malrotation may impair vascular supply, predisposing the bowel to ischemic injury and subsequent atresia. This pathogenesis was proposed by Fishman et al. [
3], who reported 10 cases of colonic atresia, including 3 cases with concurrent malrotation and Hirschsprung’s disease. They emphasized that the presence of a non-fixated colon in a newborn with colonic atresia should prompt suspicion of Hirschsprung’s disease in the distal segment and performing a biopsy of the distal colonic segment at the time of surgery is a prudent approach. Emerging evidence also implicates genetic factors, such as mutations in RET or EDNRB pathways, which regulate enteric neural development and vascularization [
1,
4].
The simultaneous occurrence of esophageal atresia, Hirschsprung’s disease, and intestinal atresia, as seen in Case 1, is exceedingly rare and present diagnostic and therapeutic challenges. To the best of our knowledge, there has only been one case of coexistence of esophageal atresia, intestinal atresia, and Hirschsprung’s disease reported in the literature [
7]. The interplay of these anomalies can obscure clinical presentation and delay definitive diagnosis. As seen in Case 1, esophageal atresia may initially mask the symptoms of Hirschsprung’s disease or intestinal atresia until surgical exploration reveals these anomalies. The diagnostic complexity is further compounded by rare findings, such as ganglion cells distal to the site of intestinal atresia, which challenge conventional understanding of aganglionosis. Early and accurate diagnosis requires a high index of suspicion, especially when postoperative outcomes deviate from expectations [
7,
8].
The clinical management of these cases highlights the need for a multidisciplinary approach. Surgical interventions must be carefully staged to address each anomaly while minimizing risks. Surgical techniques, such as temporary fecal diversion or staged pull-through procedures, have proven effective in stabilizing patients before definitive repair. Understanding the embryologic and pathophysiologic connections between these anomalies not only aids in early detection but also opens avenues for exploring genetic and environmental factors that might predispose to such rare and complex phenotypes [
7,
8].
To our knowledge, this is one of the few reports describing the coexistence of colonic atresia, malrotation, and Hirschsprung’s disease with histologic confirmation. Unlike previous studies, our report highlights the importance of considering aganglionosis when colonic atresia is accompanied by a non-fixated colon, emphasizing the necessity of intraoperative biopsy and surgical planning tailored for staged rectorectal pull-through procedures.
In conclusion, colonic atresia combined with malrotation and Hirschsprung’s disease is rare, and timely diagnosis and management is challenging. Careful review of the clinical course and high level of suspicion is necessary for timely management.
NOTES
-
Conflict of Interest
No potential conflict of interest relevant to this article was reported.
-
Author Contributions
Conceptualization: L.S.; Data curation: P.S.; Investigation: L.S., P.S.; Methodology: P.S.; Project administration: L.S., S.J.M.; Resources: S.J.M.; Software: K.W.; Supervision: P.S.; Visualization: K.W.; Writing - original draft: P.S.; Writing - review & editing: P.S., L.S.
Fig. 1.Intraoperative findings from Case 1 showing colonic atresia and non-fixated distal colon suggestive of malrotation.
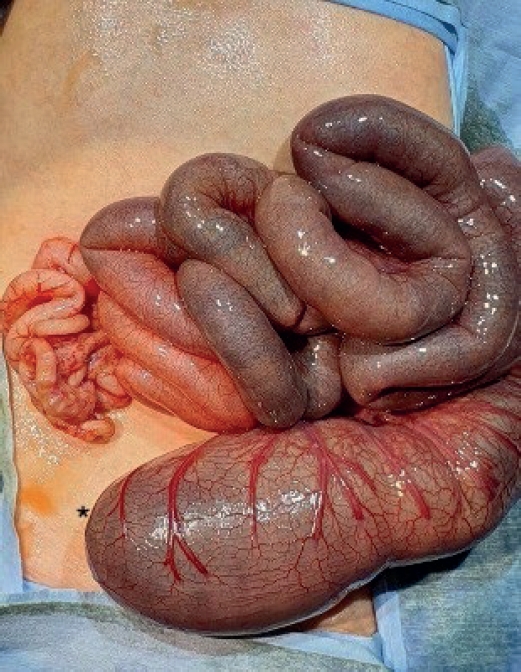
Fig. 2.Intraoperative findings from Case 2 showing colonic atresia with malrotation.
REFERENCES
- 1. Lauwers P, Moens E, Wustenberghs K, Deprettere A, Ruppert M, Balliu L, et al. Association of colonic atresia and Hirschsprung’s disease in the newborn: report of a new case and review of the literature. Pediatr Surg Int 2006;22:277-81.
- 2. Janik JP, Wayne ER, Janik JS, Price MR. Ileal atresia with total colonic aganglionosis. J Pediatr Surg 1997;32:1502-3.
- 3. Fishman SJ, Islam S, Buonomo C, Nurko S. Nonfixation of an atretic colon predicts Hirschsprung’s disease. J Pediatr Surg 2001;36:202-4.
- 4. Draus JM Jr, Maxfield CM, Bond SJ. Hirschsprung’s disease in an infant with colonic atresia and normal fixation of the distal colon. J Pediatr Surg 2007;42:e5-8.
- 5. Daher P, Raffoul L, Riachy E, Mitri R. Uncommon co-occurrence of ileal atresia and total colonic aganglionosis in two unrelated newborns. Pediatr Surg Int 2012;28:85-7.
- 6. Wisbach GG, Vazquez WD. Ileal atresia, malrotation and Hirschsprung’s disease: a case report. J Pediatr Surg Case Rep 2013;1:e3-5.
- 7. Santos LP, Coimbra D, Cunha C, Lopes MF. Oesophageal atresia with tracheo-oesophageal fistula, ileal atresia and Hirschsprung’s disease: outcome of a rare phenotype. BMJ Case Rep 2019;12:e226675.
- 8. Knod JL, Bondoc AJ, Garrison AP, Bischoff A, Dickie B, Frischer JS. Concurrent esophageal atresia with tracheoesophageal fistula and Hirschsprung disease. J Pediatr Surg Case Rep 2015;3:499-500.
Citations
Citations to this article as recorded by