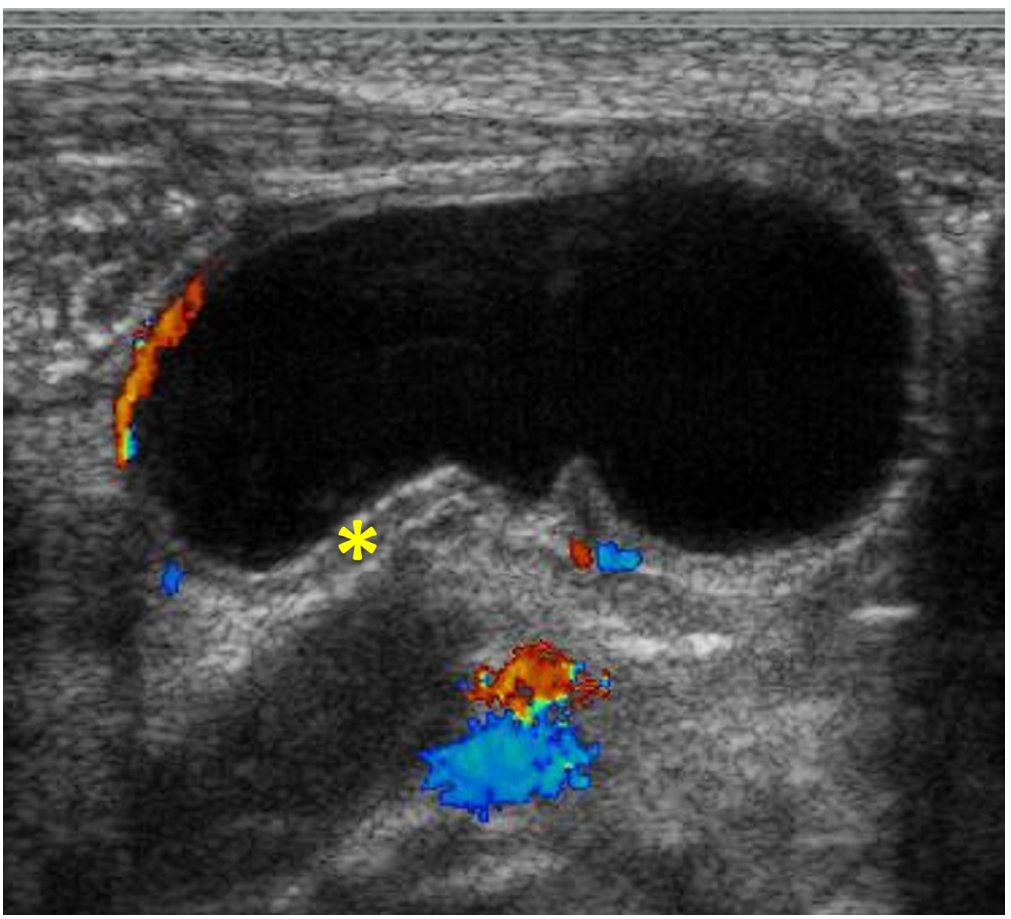
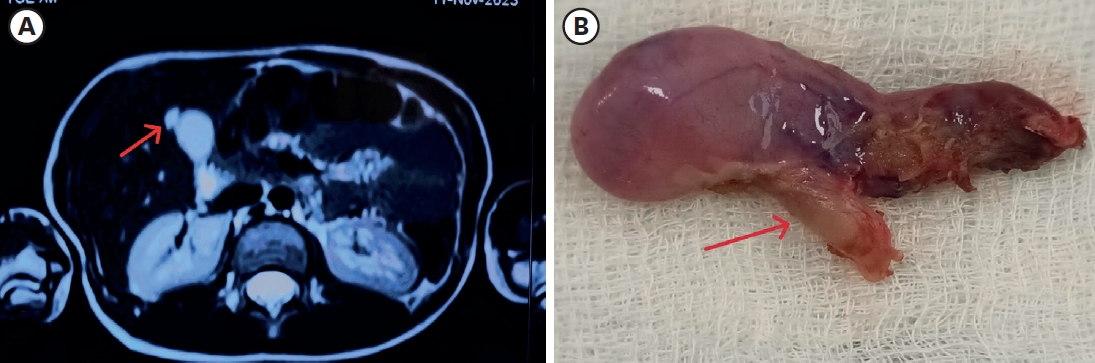
Anorectal duplications account for only 5% of gastrointestinal duplications, and cases with involvement of the anal canal are much rarer. Nearly all anorectal duplications are posterior to the rectum; duplications located anterior to the normal rectum are highly unusual, and only a few cases have been reported. We report the case of an anterior anorectocolonic duplication presenting as a rectovaginal fistula in a 2-month-old infant. After diagnosis, the duplication was excised completely without further intestinal complications.
We report a neonatal case of “intraluminal” pyloric duplication cyst, causing gastric obstruction after birth. Endoscopy revealed a submucosal cystic lesion approximately 15 mm in size arising from the anterior and inferior surfaces of the pylorus obliterating the pyloric canal. After laparotomy, intraoperative cholangiography was performed, which documented no communication between the cyst and the bilio-pancreatic duct. Gastrotomy was performed transversally over the antrum, and the cyst delivered through the incision. The cyst was incised, the upper part of the cyst wall removed, and a mucosectomy performed on the inner cyst wall of the lower part. The mucosa and muscle of the margin of the cyst were approximated. At follow up of 10 months, the patient is well without any sign of gastric obstruction.
Citations
The duplication of gastrointestinal tract has been known to be a rare condition and two different forms, cystic and tubular type. This study was conducted to examine its clinical characteristics, especially cystic enteric duplication which was detected antenatally or postnatally.
There were 13 patients, who confirmed as cystic enteric duplication after operation between July 1996 and June 2015. Clinical data, including a gender, age at operation, presenting symptoms, diagnostic modalities, locations of lesion, and results of surgical treatment, were reviewed retrospectively according to cases detected antenatally and postnatally.
Five cases were included in antenatal diagnosis group and 8 cases in postnatal diagnosis group. Both groups show slightly common in female and the lesion most common in ileum. Antenatal diagnosis group shows 2 males and 3 females and the mean age at operation was 12±52 days (range, 5 to 90 days). They received operation regardless of symptom. Postnatal group shows 3 males and 5 females and the mean age at operation was 462.5±777.0 days (range, 4 days to 6 years). Moreover, 6 patients (75.0%) were age before 2 years. They usually presented abdominal pain with vomiting.
Cystic enteric duplication could present symptoms at any time during childhood, mainly before 2 years old, and so a proper management should be considered when suspect it. Although it is uncommon, surgical management including a minimal invasive procedure could be attempted despite the neonatal period.
Gastric duplication is a rare anomaly which account for only 3.8% of all gastrointestinal duplication. Gastric duplications are usually cysticlesion without communication with lumen. Most frequent presentation is an abdominal mass with vomiting, mainly diagnosed within the first year of life. Surgical removal is necessary in all cases, and optimal timing for surgery is the time that diagnosis is made. However, prenatally diagnosed gastric duplication is getting more common, and determining timing for surgery is not easy due to absent or minimal symptoms just after birth. We experienced prenatally diagnosed gastric duplication in a female newborn baby that gastric duplication was suggested in 24th week of gestational age through prenatal ultrasonogram. Surgical removal was done at 3 months after birth, and showed good results. We think that natural history of gastric duplication and prevalent age of surgical disease which is similar to gastric duplication such infantile hypertrophic pyloric stenosis should be considered when timing of surgery on prenatally gastric duplication is decided.
Intestinal duplication is a rare congenital anomaly. The diagnostic approach is difficult because of the differences in its location and clinical presentation. To evaluate the diagnostic as well as the therapeutic approaches in children, the medical records of 20 patients with intestinal duplications which had been operated upon from July 1980 to October 2002 were analyzed, retrospectively. The range of age was from 1 day to 11 years. The variables, such as age, sex, clinical presentation, diagnostic method, localization, anatomic type, treatment, complication, and combined anomalies were analyzed. Most of the cases were presented as incidental finding. The majority of the duplications except hindgut were cystic type. Treatment included segmental intestinal resection, excision of the lesion without intestinal resection, and septotomy. Seventy-five percent of the patients were detected before 1 year of age. The anatomic type of the lesion was closely related with its location. The cases of hindgut were almost always tubular type except 1 case. Clinical presentation was related to age, location, and anatomic type. There were no specific diagnostic methods. Perfect localization and application of appropriate operation are the most important requirements for successful treatment.
Citations
Anal canal duplications occurring in a pair of 4 month-old healthy female twins are presented. The openings were located in the posterior midline of the anus since birth without a history of perianal abscess or swelling. Excision of the duplicated anal canals was performed using posterior sagittal approach. Although the anal canal duplication occurs predominantly in female, to our knowledge, this is the first case of anal duplication in a monozygotic female twins reported.
Gastrointestinal duplications are rare congenital malformation that may require surgical intervention in the neonate, infant, and occasionally the older child. Symptoms produced by duplications vary according to their location, size, type and histology. We report the clinical characteristics and the surgical results of 9 cases of the gastrointestinal duplications treated at at Asan Medical Center between 1989 and 2000. Five patients were boys and four were girls; age of patients ranged from 5 days to 10 years. Eight duplications were cystic and one was tubular. One involved the stomach; five were in the ileum, and two in the cecum. The most common presentation was intestinal obstruction. There was associated anomaly in one patient, pulmonary sequestration and double ureter. Ectopic gastric mucosa was found in two. All patients underwent surgical resection. There was no perioperative mortality or morbidity. Although gastrointestinal duplication is a rare entity, consideration of associated anomalies and being familiar with the anatomy and clinical features are required for adequate management. In cystic form, complete excision is recommended but planned surgery is required for long segment tubular lesion.
Citations
Segmental dilatation of small intestine is a rare form of the congenital intestinal anomaly. Many other congenital anomalies have been reported in these patients, but to our knowledge, the association with colonic duplication has not been reported in literatures. We report a case of segmental dilatation of distal ileum associated with colonic duplication. The main clinical and pathogenic aspects are discussed, and the literatures were reviewed.
Congenital duplication of the gallbladder, including true duplication and septated gallbladder, is an uncommon but potentially complicated malformation. It presents biliary colic associated with acute or chronic cholecystitis. It can be diagnosed preoperatively by various imaging tools such as real-time ultrasonography, biliary scintigraphy, oral cholecystography, or endoscopic retrograde cholangiography. It can be safely managed by either laparoscopic or open procedures. This report describes a 6-year-old girl with true duplication of gallbladder diagnosed preoperatively by real-time ultrasonography and treated by open cholecystectomy.
Diphallus is a rare congenital anomaly and is frequently associated with duplication of the urinary tract and rectosigmoid, and commonly associated with vertebral anomalies. Remzi reported less than 100 cases of duplication of all or a portion of the penis, but about 10 cases of complete diphallus with exstrophy of cloaca was reported, and a case of complete diphallus associated with hingut duplication was reported, and complete diphallus with displacement of bladder associated hindgut duplication and imperforate anus was not reported in Korea. We experienced a case of the complete diphallus associated with displacement of bladder, hindgut duplication, and imperforate anus as a variant of cloacal exstrophy. A review of published cases suggests that this may be the first example of a complete dip hall us with displacement of bladder coexisting with the hindgut duplication and imperforate anus.
 First
First Prev
Prev