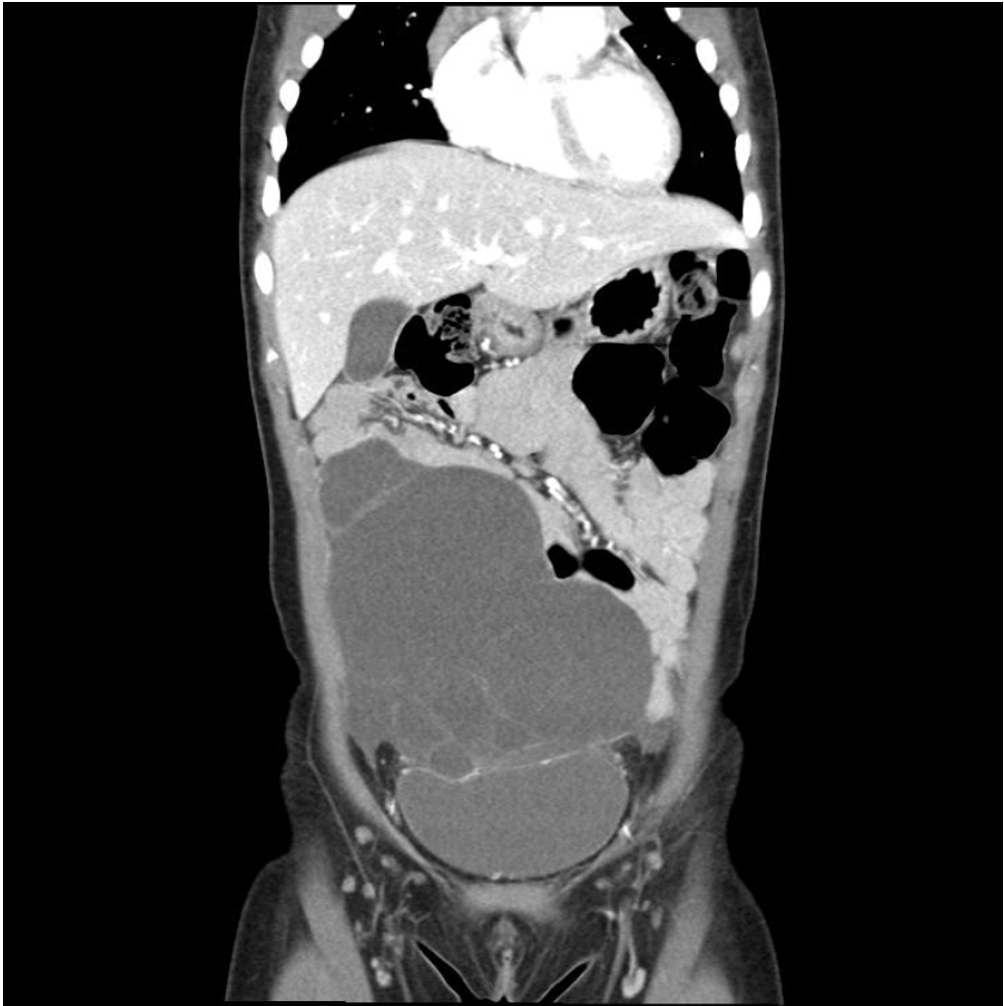
We evaluated perioperative and long-term outcomes of minimally invasive surgery (MIS) and established indications of MIS in solid pseudopapillary tumor (SPT) in pediatric patients.
From October 1992 to April 2018, 66 patients (age, <18 years) diagnosed with SPT underwent either open pancreatectomy (OP) or MIS. Variables including postoperative complications and recurrence rates were retrospectively analyzed.
Thirty-five patients underwent open surgery and 31 underwent laparoscopic/robotic surgery. Mean tumor size in MIS was significantly smaller than that in OP (4.3±1.8 cm vs. 7.6±3.5 cm, p=0.005). There were 4 cases of open conversion from laparoscopic surgery because of vessel encasements (n=2), bleeding (n=1), and pancreatic ductal injury (n=1). Solitary pseudopapillary carcinoma was diagnosed in 6 patients. Recurrence was observed in 3 and 1 patients who underwent OP and MIS, respectively (p=0.634). Tumor size, mass size/abdominal diameter (MS/AD) ratio, and degree of the portal or superior mesenteric vein involvement were the most important indications for MIS.
MIS is being widely used in pediatric surgeries with increased expertise and safety, especially in pancreatic diseases. Careful patient selection for MIS in regards with parameters such as MS/AD ratio and vessel abutment might be a feasible choice.
Citations
Polypoid or tumorous arteriovenous malformation (AVM) of small intestine is rare and can be misdiagnosed as other tumorous conditions. We experienced a rare case of giant jejunal AVM in a 15-year-old boy, who complained of intense abdominal pain. Ultrasonography and contrast-enhanced CT revealed a 13.5-cm-sized multiseptated cystic mass arising in small intestine, which was mimicking submucosal tumor. It was successfully treated by surgical resection. The specimen showed a multilobulated outbulging submucosal mass in jejunum. Histopathologic evalulation confirmed AVM located in the submucosa, muscularis propria and subserosa. This case is the largest AVM of small intestine among which has ever been reported.
Malignant ovarian tumors in children are very rare, and consist of about 1% of all childhood malignant tumors. The purpose of this study is to examine the clinical characteristics, treatment, and prognosis for children with malignant ovarian tumors. We retrospectively reviewed the medical records of children under 15 years of age with malignant ovarian tumors who had been treated surgically at Asan Medical Center between 1989 and March 2009. There were 32 patients, ranged in age at surgery from 2 to 15 years (mean; 10.4 years). The median follow-up period was 64.7 months (from 1 month to 188 months). Pathologic diagnosis were; immature teratoma (n=10), mixed germ cell tumor (n=10), and dysgerminoma (n=6). Tumor stage was classified by the staging system of the International Federation of Gynecology and Obstetrics (FIGO). The number of patients in stage I, II, III, and IV were 24 (75%), 2 (6.2%), 4 (12.5%), and 2 (6.1%), respectively. The tumor recurred in 4 patients. Seven patients of group 1 did not receive postoperative adjuvant chemotherapy, and in three of them, the tumor recurred. Twenty-five patients (group 2) underwent postoperative adjuvant chemotherapy, and there was only one recurrence. One patient who did not receive postoperative adjuvant chemotherapy and expired 10 months after operation because of tumor recurrence and distant metastasis. The overall 5-year event free survival (EFS) was 84.2%: group 1 in 44.4%, and group 2 in 95.7%. Tumor recurrence was related to the postoperative adjuvant chemotherapy (p=0.004). In conclusion, proper surgical procedures with relevant postoperative adjuvant chemotherapy might improve clinical results in children with malignant ovarian tumors.
Pancreatic tumors in children are relatively rare, and their prognosis differs from that in adults. The purpose of this study is to examine the clinical characteristics, treatment, and prognosis for children with pancreatic tumors. We retrospectively reviewed the medical records of children under 15 years of age with pancreatic tumors who were treated surgically at Asan Medical Center between January 1992 and November 2009. There were 16 patients, fourteen of whom were pathologically diagnosed with solid pseudopapillary tumor. The other two patients were diagnosed with pancreatoblastoma and acinar cell carcinoma, respectively. Six patients of the 16 patients (38%) were male, and there was a male-to-female ratio of 1:1.6. The initial presentations were upper abdominal pain in eight patients (50%), palpable abdominal mass in three, and vomiting in one. Four patients were diagnosed incidentally. Six patients' tumors were located in the pancreatic head, six in the pancreatic body, and four in the pancreatic tail, respectively. The surgical procedures performed included distal pancreatectomy (n=7, 44%), median segmentectomy (n=3), enucleation (n=3), pancreaticoduodenectomy (n=2), and pylorus-preserving pancreaticoduodenectomy (n=1). Three patients underwent laparoscopic surgery. The median tumor size was 6.5cm (1.8~20 cm). Early surgical complications included pancreatic fistula (n=4), bile leakage (n=1), and delayed gastric emptying (n=1). A late complication in one patient was diabetes. The median follow-up period was five years and four months, and all patients survived without recurrence. While pancreatic tumors in adults have a poor prognosis, pancreatic tumors of childhood are usually curative with complete resection and thus have a favorable prognosis.
We report a case of nonfunctioning neuroendocrine tumor of the pancreas in a 15-year-old girl who presented with back pain. On physical examination, there was mild tenderness in the left upper quadrant of the abdomen. The patient had no pancreatic hormone-associated symptoms. An abdominal ultrasonography showed a well-demarcated hypervascular solid mass with calcification in the tail of the pancreas. An abdominal computed tomography scan showed a 6x5cm sized well-encapsulated enhancing solid mass with cystic component in the tail of the pancreas. Distal pancreatectomy was performed. Pathology revealed awell- differentiated nonfunctioning low grade malignant neuroendocrine tumor of the pancreas. The postoperative course was uneventful.
Sertoli-Leydig cell tumor is a rare sex-cord stromal tumor of the ovary. They make up less than 0.5 % of all ovarian tumors. We experienced a case of an ovarian Sertoli-Leydig cell tumor in a 4 year-old girl who presented with nausea, vomiting, and lower abdominal pain of 2 days' duration. On physical examination, there was mild tenderness in the right lower quadrant of the abdomen. Abdominal ultrasonography and computed tomography (CT) scan revealed a pelvic mass measuring 5 × 3 cm that appeared to arise from the right ovary. At exploratory laparotomy, a 6 × 5 × 3 cm solid right ovarian mass without torsion was found. A right salpingo-oophorectomy was performed. The postoperative course was uneventful. The child was discharged 5 days after surgery.
Citations
Inflammatory myofibroblastic tumor (IMT) is a rare reactive lesion characterized by the feature of myofibroblasts and a mixed inflammatory infiltrate that rarely undergoes malignant transformation. Extrapulmonary IMTs in children have been described involving the mesentery, omentum, retroperitoneum, abdominal soft tissues, liver, bladder, mediastinum, head and neck, extremity, appendix, and kidney. Medical records of children treated with abdominal IMT between 1985 and 2005 were reviewed retrospectively. Seven children were treated for IMT with the mean age of 3y 2m (range, 1y 1m to 14y). Tumors were located in transverse mesocolon (n=2), omentum (n=1), porta hepatis (n=2), complex site (antrum, duodenum, common bile duct, porta hepatis) (n=2). The symptoms included abdominal mass, fever, jaundice, abdominal pain and anemia. The masses were excised totally in transverse mesocolon, omentum IMT and there is no evidence of recurrence (follow-up periods: 6y 8m, 8y 9m, 4y 10m). In porta hepatis IMT, liver transplantations were performed and there is no evidence of recurrence (follow period: 6y 8m, 8y 7m). In one case of complex site IMT, partial excision of mass was performed and he still survived with no change of the residual tumor during follow-up period. The other one of complex site IMT denied further treatment after the biopsy. In conclusion, complete surgical excision including liver transplantation and close follow-up are mandatory for the abdominal IMT in child.
Citations
Pancreatic tumors in children are very rare but have a better prognosis compared with that in adult. Pediatric pancreatic tumors are more often benign and easier to resect. To evaluate the characteristics and prognosis, the records of 13 patients who underwent pancreatic resection, from June 1997 to May 2005, at Samsung Medical Center were reviewed. The mean follow up period was 48 months. The male to female ratio was 1: 1.6. Mean age was 10.3 years. Signs and symptoms included abdominal pain (7), abdominal palpable mass (5), jaundice (1), hypoglycemic (1), and non-specific GI symptoms (4). The commonly used diagnostic tools were CT and abdominal sonography. In addition, MRI, ERCP, EEG, and hormone test were also done when indicated. Surgical procedures included distal pancreatectomy (5), pylorus preserving pancreaticoduodenectomy (4), tumor excision (3), and subtotal pancreatectomy (1). Locations of lesions in pancreas were head (4), tail (5), and body and tail (4). Postoperative complications developed in 3 cases; postoperative ileus (1), wound problem (1), and pancreatitis (1). The pathologic diagnosis included solid-pseudopapillary tumor (6), congenital simple cyst (1), pancreatic duplication cyst (1), serous oligocystic adenoma (1), mucinous cystadenocarcinoma (1), rhabdomyosarcoma (1), insulinoma (1), and pancreatoblastoma (1). Three cases received adjuvant chemotherapy and radiotherapy. Overall survival rate was 81%. One patient with a mucinous cystadenocarcinoma died. In this study, pancreatic tumors in children were resectable in all patients and had good survival. Surgery of pancreatic tumors should be regarded as the gold standard of treatment and a good prognosis can be anticipated in most cases of benign and malignant tumors.
Liver tumors in children are rare, relatively complex, and encompass a broad spectrum of disease processes. This study reviews our experience of liver tumors during the last 10 years. Medical records of 36 cases of liver tumors?in children, treated at Samsung Medical Centers, from October 1994 to December 2005, were reviewed in this study. We analyzed disease characters and survival rates as a whole and by specific disease. The median age was 3.6 years. Male and female ratio was 1:1. The most common symptom was the palpable mass in 15 cases. Others were abdominal distension in 9 cases, jaundice in 2, vomiting in 2, weight loss in 2, and pubic hair growth in 1. CT or US and liver biopsy were performed for diagnosis. There were 28 malignant tumors: malignant rhabdoid tumor (1 case), hepatocellular carcinoma (3 cases), hemangioendothelioma type II (3 cases), angiosarcoma (1 case), and hepatoblastoma (20 cases). Eight tumors were benign; hepatic adenoma (1 case), focal nodular hyperplasia (2 cases), hemangioendothelioma type I (2 cases), mesenchymal hamartoma (3 cases). In this study the clinical characteristics were not different from the other reports. Liver transplantation was performed in 3 cases-1 with hepatoblastoma and 2 with hepatocelleular carcinoma. Accurate and early diagnosis, and individualized multi- modality therapeutic approaches might be important for better outcome.
Citations
Solid pseudopapillary tumor (SPT) of the pancreas occurs most frequently in the second or third decades of life, and is prevalent in females. Unlike other pancreatic malignancy, SPT usually has a low malignancy potential. This study reviews our clinical experience and surgical treatment of pancreatic SPT. Admission records and follow-up data were analyzed retrospectively for the period between January 1996 and January 2003. Five patients with a pancreatic mass were operated upon and SPT was confirmed by pathology in each case. The male to female ratio was 1 : 4. The median age was 13.8 years. Findings were vague upper abdominal pain (n=5, 100 %) and an abdominal palpable mass (n=3, 60 %). The median tumor diameter was 6.8cm and the locations were 2 in the pancreatic head (40 %) and 3 in the pancreatic tail (60 %). Extra-pancreatic invasion or distant metastasis was not found at the initial operation in all five cases. A pyloruspreserving pancreaticoduodenectomy (n=1) and a mass enucleation (n=1) were performed in two patients of pancreatic head tumors. For three cases of tumors in pancreatic tail, distal pancreatectomy (n=2) and combined distal pancreatectomy and splenectomy (n=1) were performed. The median follow-up period was 60 months(12-117month). During the follow-up period, there was no local recurrence, nor distant metastasis. Postoperative adjuvant chemotherapy or radiotherapy was not carried out. All five children were alive during the follow up period without any evidence of disease relapse. SPT of the pancreas in childhood has good prognosis and surgical resection of the tumor is usually curative.
Primary liver tumors are uncommon in childhood, with a relative frequency of 3% of childhood tumors. Seventy three cases of pediatric primary liver tumors which were operated on at single institution between 1986 and 2002 were reviewed. There were 37 cases of hepatoblastoma, 11 hepatocellular carcinomas, 6 undifferentiated (embryonal) sarcomas, and 1 mixed germ-cell tumor in malignancies. Benign tumors constitute only 24.6% of liver tumors, including 7 hemangioendotheliomas, 5 mesenchymal hamartomas, 3 congenital cysts, and one each with focal nodular hyperplasia and hemangioma. The common presenting clinical features were abdominal mass or abdominal distension. Anatomical hepatic resections were carried out in 38 cases, and non anatomical resections in 34 cases. One patient died of a direct result of hepatic resection(1.4%), and complication rate was 16.4%.
Solid pseudopapillary tumor of pancreas in children is a tumor with low malignant potentiality, and is rarely associated with distant metastasis. A 13-year-old girl was hospitalized because of abdominal pain of one week duration. Abdominal CT revealed not only a 12×6cm sized mass at the pancreatic body and tail but also a 1cm sized mass in left lobe of the liver. The patient underwent a near-total pancreatectomy and tumorectomy of the liver. A solid pseudopapillary tumor with liver metastasis was confirmed by pathology. She has undergone 13 courses of chemotherapy and has been well for 13 months without any sign of recurrence.
Neuroblastoma arises from the embryonic tissue of the adrenergic rest. It is commonly found in children and mostly in nonrenal tissue. We present a case of intrarenal neuroblastoma which was initially thought to be a Wilms' tumor. The patient was a 18 months-old girl treated with radical nephrectomy and adjuvant chemotherapy after operation. The neoplasm within the kidney in children cannot always indicate Wilms' tumor. Neuroblastoma of the adrenal gland or retroperitoneal tissue may often compress or invade the kidney directly or arise from the kidney. Clinical aspects that differentiate between neuroblastoma and Wilms' tumor are discussed with a review of the literatures.
Pediatric solid tumors have many similarity among different tumors. These tumors present small round cell types, and cause frequent diagnostic problems in pediatric pathology. An important advance in the investigation of these small round cell tumors has been the identification of consistent chromosomal translocations associated with several types of tumor. Eighteen patients with soft tissue sarcoma were available for review. Seventeen cell lines were also included in this study. The RNA from the specimens were analyzed by reverse transcriptase-polymerase chain reaction (RT-PCR). PAX3-FKHR fusion was present in four of five alveolar rhabdomyosarcoma and PAX7-FKHR fusion was detected in one of five alveolar rhabdomyosarcoma. None of the specimens expressed more than one chimeric transcript. EWS-FLI1 or EWS-ERG fusions were detected in all seven Ewings' sarcoma. No specimens showed EWS-WT1 fusion. These results corresponded well to the histopathologic diagnosis. There were no differences in the histologic appearances of tumors with the more frequent PAX3-FKHR or EWS-FLI1 fusions compared with those containing the variant PAX7-FKHR or EWS-ERG fusions. RT-PCR assay for chimeric transcript is an useful tool for a rapid and
objective
diagnosis of pediatric solid tumors. Through these tools, we can approach genetically to the differential diagnosis of undifferentiated small round tumors.
Among 60 children with teratoma, forty-three (71.7 percent) were girls and 17 (28.3 percent) boys. Primary sites were sacrococcygeal in 30 patients (50 percent), retroperitoneal in 12 (20 percent), ovarian in 11 (18.3 percent), testicular in 3 (5 percent), and one in each of nasopharyngeal, gastric, hepatic and pancreatic (1.6 percent, respectively). Fifty-five (91.7 percent) teratomas were benign and 5 (8.3 percent) malignant. Malignant teratomas were detected only at sacrococcygeal region (16.7 percent). Older than 2 months of age at diagnosis, presence of urinary and colonic obstructive symptoms, multiple masses and elevated serum alpha-fetoprotein were indicators of malignancy in sacrococcygeal region. Tumor size, presence of calcification, and gross appearance (cystic or solid) did not correlate with malignant nature. Thirteen (21.7 percent) cases were associated with other anomalies. For the immature teratoma, the operative resection without adjuvant chemotherapy was enough. Three malignant cases were survived, one with chemotheapy for 3 years and the others without chemotherapy for 5 and 10 years.
A 3-year-old boy with a Wilms' tumor had unusually severe hypertension, polydipsia, polyuria and hypokalemia. Physical-examination on admission was unremarkable except for the presence of a smooth, firm mass in the right abdomen. Computerized tomography showed a tumor occupying the upper two thirds of the right kidney. Plasma renin activity and aldosterone concentration were markedly elevated, 37.7 mg/ml/hour(normal in supine position 0.15-2.33 mg/ml/hour) and 120.1 ng/dL(normal in supine position 1 to 16 ng/dL) , respectively. Hypertension varied from 150/90 mmHg to 240/180 mmHg, and was not effectively controlled by antihypertensive drugs. Right nephrectomy was performed on the sixth hospital day. At laparatomy, there was no evidence of mechenical compression of the renal artery by the tumor. The tumor, about 8 cm in diameter, was confined to the renal capsule without involvement of the renal blood vessels at the hilum. Histopathology was Wilms' tumor of favorable histology. On electron microscopy, tumor cells contained intracytoplasmic electron dense secreting graules, suggesting the possibility of renin secreting tumor cells. Shortly after nephrectomy, signs and symptoms were relieved dramatically, and plasma renin activity and aldosterone concentration were also decreased to normal.
We reviewed 45 cases of ovarian tUmors treated at Seoul National University Children's Hospital from 1983 to 1993. Forty-five patients were operated upon for 52 ovarian tumors. The most common pathologic diagnosis was mature teratoma. The next were functional cyst, the tumors of epithelial cell origin, and those of stromal origin in order of frequency. Six patients(13%) had malignant tumor. There were one malignant teratoma, two dysgerminomas, one endodermal sinus tumor, and two granulosa cell tumors. Four cases were diagnosed as torsion of ovarian cyst preoperatively, and emergency exploratory laparotomy were performed. There were three cases of ovarian tumors associated with precocious puberty. The most widely used diagnostic tool was ultrasonography.
In the treatment of these 45 patients, unilateral oophorectomy was done in 38 cases, unilateral oophorectomy with wedge resection of contralateral ovary was done in 5 cases, unilateral oophorectomy with contralateral simple cystectomy was done in one case and total abdominal hysterectomy with bilateral salpingooophorectomy was done in one case. Of the six cases of malignancy, five patients are alive 2 to 6 years after operation and one case was lost to be followed up.
 First
First Prev
Prev