Biliary atresia is a progressive sclerosing cholangiopathy of bile ducts. Most of the time it is an isolated anomaly but can present with syndromic forms. The biliary atresia splenic malformation (BASM) syndrome is associated with splenic anomalies, vascular anomalies, and visceral asymmetry along with biliary atresia. The surgical anatomy of BASM is distinctive and creates a challenge for the surgeons. We are describing a case of BASM with situs inversus and highlighting the approach, intraoperative anatomy, and surgical intricacies of Kasai portoenterostomy in such a situation with the review of pertinent literature.
Biliary atresia is a progressive sclerosing cholangiopathy of bile ducts. Most of the time it is an isolated anomaly but can present with syndromic forms. The biliary atresia splenic malformation (BASM) syndrome is associated with splenic anomalies, vascular anomalies, and visceral asymmetry along with biliary atresia. The surgical anatomy of BASM is distinctive and creates a challenge for the surgeons. We are describing a case of BASM with situs inversus and highlighting the approach, intraoperative anatomy, and surgical intricacies of Kasai portoenterostomy in such a situation with the review of pertinent literature.
Biliary atresia (BA) is a progressive sclerosing cholangiopathy of bile ducts (extrahepatic as well as intrahepatic) and its incidence varies between 1/9000 to 1/15000 [1]. The Japanese classification divides BA into 3 types depending upon the proximal-most level of bile duct atresia. Type 3 is the most common, where proximal atresia extends up to the porta. Another classification proposed by Davenport divides BA into 4 clinical groups; Syndromic BA, Cystic BA, CMV-associated BA, and Isolated BA [2].
The biliary atresia splenic malformation (BASM) syndrome encompasses BA, splenic anomalies (usually polysplenia), vascular anomalies (usually preduodenal portal vein and absence of vena cava), visceral asymmetry (usually situs inversus), and cardiac anomalies [2, 3, 4]. The abnormal, misoriented, and variable anatomy in cases of BASM always poses a challenge for the surgeon. Here, we are discussing a case of BASM with its intraoperative findings and surgical intricacies.
A 58 days old female child (weight=2 kg) presented with the complaint of clay-colored stool since birth and appearance of jaundice from day 5 of life. The child was one of the twin babies delivered via cesarean section at 34 weeks, with a birth weight of 1.9 kg. The other baby was a male child with a birth weight of 2.1 kg and he had no complaints at all. On clinical examination, the child was icteric and the liver was palpable. On investigations, liver function tests (LFT) were suggestive of high direct bilirubin (12.1 mg/dL) while alanine aminotransferase (ALT), aspartate aminotransferase (AST), and gamma-glutamyl transferase (GGT) were normal. The ultrasonography and 2D echo were suggestive of dextrocardia and situs inversus with a small-contracted gall bladder (GB). The hepatobiliary iminodiacetic acid (HIDA) scan was suggestive of the absence of bilio-enteric drainage.
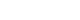
The child was taken for an intraoperative cholangiogram which suggested the absence of the extrahepatic bile ducts and a decision for Kasai portoenterostomy (KPE) was taken. The left subcostal incision was extended towards the midline and to the right. On exploration, the liver was dark-colored, cirrhotic, and firm (Fig. 1A), polysplenia was present over the right side (Fig. 1B), the stomach was on the right side (Fig. 1C), duodenojejunal (DJ) flexure was in the midline and the ileocecal junction was free lying. The GB was dissected from its bed and dissection was done towards the porta hepatis (Fig. 2A). The proper hepatic artery and its bifurcation were identified, and they were overlapping the portal vein bifurcation (Fig. 2B). The portal plate was dissected in between the portal vein bifurcation (Fig. 2B), Roux loop of the jejunum (20 cm) was created 15 cm from DJ flexure and it was brought towards the porta. Portoenterostomy was done with interrupted delayed absorbable 5-0 sutures (Fig. 2C) and a liver biopsy was taken.
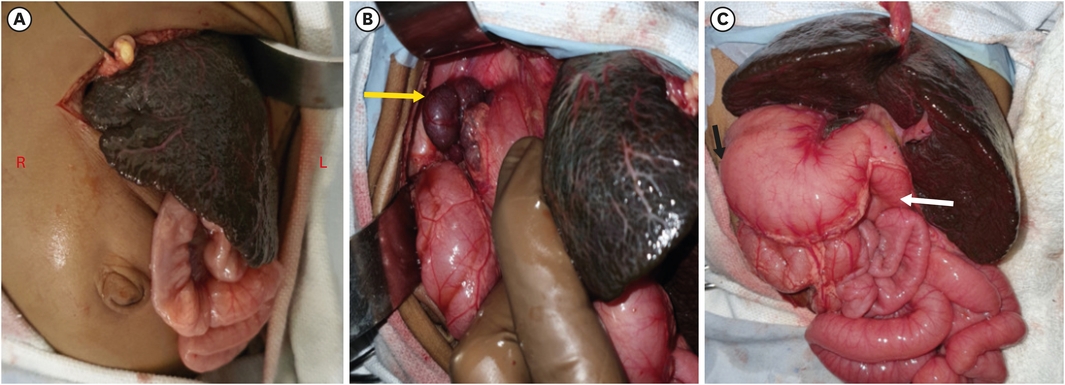
Fig. 1
Showing appearance (color and neo capillaries) of the liver and left sided incision (A), polysplenia (B) on the right side (yellow arrow) of the abdomen, greater curvature (black arrow) towards right and the duodeno-jejunal flexure (white arrow) in the midline.
R, right side; L, left side.
Fig. 2
Showing dissection at the porta (A and B), portal plate area (white arrow) surrounded by the hepatic artery bifurcation (red arrow) and the portal vein (blue arrow) with completed portoenterostomy (C), and the roux limb (red star) in between the duodenum (blue star) and the transverse colon (white star).
The postoperative course was smooth and the child was passing green color stools on day 7 and got discharged on day 8 with an oral antibiotic, ursodeoxycholic acid (UDCA), phenobarbitone, and multivitamin supplementation.
During follow-up after one month child was active, tolerating feed well, non-icteric but complaining of passing acholic stools intermittently. Steroid (prednisolone 2 mg/kg/day) was added for 1 month then tapered and stopped. At 3 months follow-up, the child was fine, passing normal color stools and LFT was suggestive; total bilirubin=2.2 mg/dL, normal AST/ALT, and GGT.
The incidence, associated abnormalities, and prognosis of BASM vary in different reported studies. In a retrospective study of 28 years, Davenport et al reported a 10.2% incidence of BASM and a poor prognosis of BASM than isolated BA [3]. He hypothesized that these infants have intrinsically worse liver disease or at least fewer exposed biliary ductules at the transacted portal plate during KPE. While Zhan et al done a study in china over 851 patients with BA and reported a 4.9% incidence of associated anomalies with cardiac anomalies were the most common and the prognosis was not different from isolated BA [4]. Gupta et al from India reported a 3.4% incidence of polysplenia and situs inversus in a study of 137 patients of BA [5].
The etiopathogenesis of BA itself remains speculative and probably is the result of a sclero-inflammatory process by an unknown etiology that leads to the complete obstruction or absence of the extrahepatic bile ducts. The BASM may have a different etiopathogenesis because in this entity various other organs are also involved which suggests that the insult happened at around the fifth week of gestation during the phase of organ development [6]. The early embryonic pathology with associated abnormalities has been mentioned as a cause of poor prognosis in these patients.
In our case, the child was premature (34 weeks) and one of the twins' babies. Durkin et al reported a relatively high incidence of BASM in premature discordant twins babies [7]. The index case presented at 58 days of life and if we calculate corrected age according to prematurity, the child was around one month only. Even then the child's liver was showing cirrhotic changes i.e., dark-colored, nodular appearance, neo capillary formation (Fig. 1A), and firm consistency. It suggests that along with the biliary duct atresia, the liver is also developmentally abnormal in BASM.
Abdominal heterotaxy, malrotation, and associated anomalies in a case of BASM pose difficulties during dissection as well as during identification of structures, it also creates a problem with the orientation of the Roux-en-Y loop. In our case, during dissection, we encountered an abnormal proper hepatic artery and its bifurcation, which were exactly overlying portal vein bifurcation (Fig. 2B) because of that dissection at the porta was difficult. Routinely, we prefer extended KPE, but in this case, it was not possible as the lateral dissection was less because of abnormally placed right and left hepatic artery over the portal vein which made porta hepatis narrow and deep (Fig. 2B). The posterior layer suturing of KPE was difficult and risky as it was deep and we had to avoid injury to the portal vein and hepatic artery. For portoenterostomy, we usually bring the Roux loop through a mesocolic window but in this case because of intestinal malrotation it was not possible, so Roux limb was placed in between the duodenum and transverse colon (Fig. 2C) and was fixed with duodenum (serosal sutures) to keep it in the proper position.
Postoperatively, the child was passing intermittent clay-colored stools, suggestive of ongoing sclerosis over the porta and proximal ductules. The parents were counseled regarding the need for liver transplants in the future, pediatric gastroenterology, as well as liver transplant team opinions, were also taken.
The liver biopsy of our case was suggestive of bridging fibrosis, chronic inflammation, and ductal plate malformation (DPM). The DPMs are characterized by the persistence of embryonic biliary structures after birth [8]. DPMs are considered to result from a lack of remodeling of the ductal plate during the fetal period. In the index case, the largest biliary ductule diameter at the porta was 300 micron and that indicates patency of good size ductule at the porta at the time of surgery. These all findings suggested, developmentally abnormal liver and bile ducts/ductule (extrahepatic as well as intrahepatic), in our index case the outcome would not be excellent and the child might need a liver transplant in near future.
In conclusion, BASM with situs inversus is a rare but well-reported entity and associated with other congenital surgical abnormalities. The heterotaxy of organs and associated abnormalities create surgical intricacies and require surgical modifications along with additional surgical intervention.
Conflict of Interest:No potential conflict of interest relevant to this article was reported.
Author Contributions:
Conceptualization: S.S.
Data curation: R.M., A.M.F.
Formal analysis: K.R.
Supervision: S.R., L.S.B.
Validation: S.R., L.S.B.
Writing - original draft: S.S.
Writing - review & editing: S.S., K.R.