Congenital central hypoventilation syndrome (CCHS) with Hirschsprung disease (HD), also known as Haddad syndrome, is an extremely rare disorder. Recent studies have identified the paired like homeobox 2b (PHOX2B) gene as the major gene involved in the development of CCHS. The syndrome is diagnosed when gene analysis confirms a mutation in the involved gene, but making an early diagnosis is difficult because of the rarity of the disease. In this study, we report the case of a newborn male with recurrent hypoventilation and bowel distension. HD was suspected on barium enema, and loop ileostomy was performed. After surgery, the abdominal symptoms gradually improved, but extubation was not possible owing to recurrent respiratory failure. These clinical manifestations were indicative of Haddad syndrome, and genetic testing confirmed the presence of a PHOX2B mutation. The patient was diagnosed with Haddad syndrome on the 11th day after birth.
Congenital central hypoventilation syndrome (CCHS) with Hirschsprung disease (HD), also known as Haddad syndrome, is an extremely rare disorder. Recent studies have identified the paired like homeobox 2b (PHOX2B) gene as the major gene involved in the development of CCHS. The syndrome is diagnosed when gene analysis confirms a mutation in the involved gene, but making an early diagnosis is difficult because of the rarity of the disease. In this study, we report the case of a newborn male with recurrent hypoventilation and bowel distension. HD was suspected on barium enema, and loop ileostomy was performed. After surgery, the abdominal symptoms gradually improved, but extubation was not possible owing to recurrent respiratory failure. These clinical manifestations were indicative of Haddad syndrome, and genetic testing confirmed the presence of a PHOX2B mutation. The patient was diagnosed with Haddad syndrome on the 11th day after birth.
Congenital central hypoventilation syndrome (CCHS) is a life-threatening disease characterized by an idiopathic failure of automatic control of breathing, especially during sleep, and a markedly impaired ventilator response to hypercarbia and hypoxemia. Hirschsprung disease (HD), also known as congenital aganglionic megacolon, is characterized by the absence of ganglion cells in any part of the bowel wall. The association between CCHS and HD was first described by Haddad in 1978, yet Haddad syndrome remains a rare disorder [1]. Previous studies claim that de novo mutations in the paired-like homeobox 2b (PHOX2B) gene are related to the pathogenesis of CCHS [2]. This disease is diagnosed through genetic testing, but an early diagnosis of Haddad syndrome is still difficult to make because of its unfamiliarity. This case report describes a neonate, who had difficulty in stopping respiratory support and severe abdominal distension. Surgery was performed after a finding of HD on imaging. Genetic analysis revealed the presence of Haddad syndrome.
A male infant was born to a healthy 36-year-old Korean mother at 39 weeks and 4 days of gestation. The infant was delivered via cesarean section at a private maternity hospital. There was no maternal history of drug exposure or other complications during pregnancy; nor was there any family history of congenital anomalies or hereditary diseases. The neonate had a birth weight of 3,900 g (90th percentile), height of 53 cm (90th percentile), and head circumference of 35.5 cm (75th–90th percentile). The baby had Apgar scores of 9 at 1 minutes, and 10 at 5 minutes. He was stable at birth, but desaturation with cyanosis was detected afterwards. He was transferred to our hospital 3 hours after birth and on admission, facial cyanosis and an oxygen saturation of 78% were observed. Coarse breathing sounds with crackles on both lung fields and symmetric subcostal retraction was found upon physical examination. Laboratory findings and neurologic examination results were normal. Initial radiographs indicated normal lung and heart findings; however, mild bowel distension was present. The breathing of the patient stabilized after intubation and mechanical ventilator treatment.
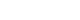
On the second day of life, abdominal distension was aggravated, and the patient was unable to pass meconium without rectal irrigation. Simple radiographs showed generalized bowel dilation, which had worsened from the previous day (Fig. 1). As the symptoms did not improve, a barium contrast enema was administered on the 4th day of life, and we checked for suspicious lesions in the transition zone of the descending colon (Fig. 2A). Warm saline irrigation was performed daily, but the abdominal distension became more severe. The patient subsequently developed a fever, and blood tests revealed an increase in C-reactive protein (CRP). Since there was no change in other findings other than the abdominal symptoms, the fever was determined to be due to an abdominal problem. The patient started taking antibiotics and we decided to skip a rectal biopsy and proceed immediately with surgery. Loop ileostomy with incidental appendectomy was performed on the 7th day of life. First, appendix resection was performed, and as a result of frozen section biopsy, it was confirmed that there were no ganglion cells. An ileostomy was performed approximately 25 cm above the ileocecal valve, and frozen section biopsy was performed at the stoma site. Some cells from the biopsy resembled ganglion cells, but they were found to require further examination. After surgery, the patient was able to pass gas and defecate through the stoma. Permanent biopsy results confirmed that the absence of ganglion cells was noted upon biopsy of the appendix and stoma site, but immunohistochemistry of calretinin and neurofilaments yielded negative results (Fig. 2B).
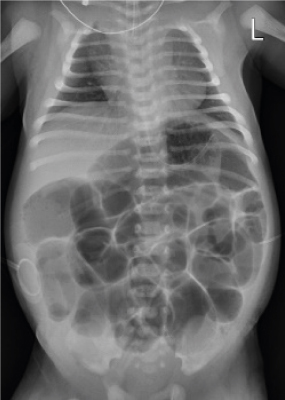
Fig. 1
Simple radiograph showing marked distension of the bowel.
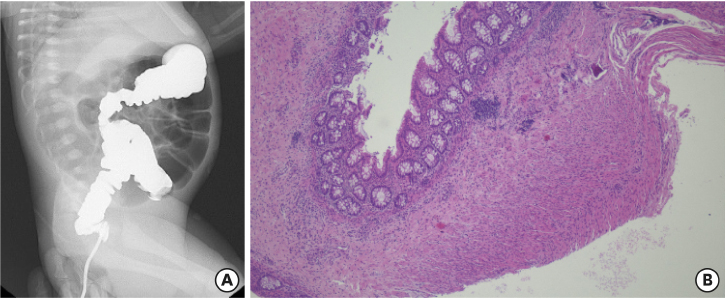
Fig. 2
Result of barium study and pathologic findings. (A) Barium enema showing a relatively narrow area of descending colon, suspicious of transition zone. (B) Absence of ganglion cells was confirmed by hematoxylin and eosin stain at the resected stoma site.
After surgery, the patient seemed to recover without lung complications, but it was impossible to disconnect the ventilator owing to recurrent apnea and hypercarbia upon extubation. Brain magnetic resonance imaging, electroencephalograph, and echocardiography were performed to identify the cause of desaturation but yielded no specific findings. Upon reviewing the patient's symptoms, clinical progression, and test results, Haddad syndrome was strongly suspected. On the 11th day of life, we performed a genetic study and confirmed the presence of a PHOX2B mutation. A heterozygote c.618dup mutation which causing a frameshift mutation in exon 3 of the gene was confirmed via Sanger sequencing (Fig. 3). It was described as NM_003924.3(PHOX2B):c.618dup in accordance with the recognized nomenclature and has been reported to be linked to CCHS in a previous study. Tracheostomy was performed, and treatment was continued at home using a mechanical ventilator. This report was exempt from Institutional Review Board (IRB) review.
Fig. 3
Sanger sequence chromatogram. Heterozygous for c.618dup (p.Ser207GlnfsTer153) on exon 3 of the PHOX2B gene was identified in the patient.
PHOX2B, paired like homeobox 2b.
CCHS is a life-threatening condition characterized by an impaired ventilatory response to hypoxia and hypercapnia, often associated with other autonomic nervous system dysfunctions. CCHS patients with HD are associated with an increased incidence of neural crest-derived tumors, such as neuroblastoma, ganglioneuroma, and ganglioneuroblastoma [3]. In a previous study, CCHS with concomitant HD was reported to be more serious than classic HD, with a high risk of having an associated tumor [4].
CCHS is a very rare disorder with an estimated incidence of 1:200,000 [5]. Clinicians suspect CCHS when other causes of central hypoventilation, such as primary pulmonary, cardiac, neuromuscular, and neurologic problems, are excluded. It is difficult to diagnose in cases of atypical presentation, or in premature infants who are often expected to have symptoms such as recurrent central apnea and feeding intolerance [6]. However, if there is a newborn suffering from persistent hypoventilation after surgical treatment of a HD, it is relatively easy to consider Haddad syndrome and an early diagnosis via genetic testing can be made.
Making diagnosis was difficult until it was revealed that the PHOX2B was important disease defined gene in CCHS. The introduction of artificial respiration was also variable and often inadequate, thus increasing the risk of neurological disorders, long-term hospitalization, and early death. CCHS patients with PHOX2B mutations were first reported by Amiel et al. [7] in 2003. Other studies have confirmed that PHOX2B mutations play an important role in the pathophysiology of CCHS [8]. The severity of CCHS patients varies widely; several studies have reported correlations between the PHOX2B genotype and CCHS phenotype. In 2015, Shimokaze et al. [9] researched the genotype-phenotype relationship of CCHS patients, with some having concomitant HD. They analyzed that most of the patients with CCHS were heterozygous for polyalanine repeat mutations (PARMs) within PHOX2B, and only about 10% have heterozygous non-PARMs (NPARMs) that include missense, nonsense, and frameshift mutations [9]. In the cases with PARMs, it has been reported that a larger polyalanine expansion was associated with worse disease severity, clinical manifestations, and prognosis [2]. Another study also suggests that patients carrying more PARMs or NPARMs present with a more severe phenotype, such as HD with extensive gut involvement, a need for continuous ventilatory support, and a higher risk for tumor development [10].
According to a case study, only one-third of patients who developed symptoms early after birth were diagnosed with CCHS within 1 month of age [9]. These results suggest that early detection is difficult owing to the rarity of the disease despite the existence of a clear method of diagnosis. Although there are still some uncertainties about the relationship between the type of genetic variation and the phenotype of the disease, it is possible to minimize sequelae when the disease is confirmed through rapid gene analysis. For CCHS patients, adequate oxygen supply and ventilation are required, and active treatment, such as tracheostomy and home mechanical ventilation; should be chosen according to the patient's condition. Appropriate treatment, done early in the disease, can lower mortality, reduce neurological damage, and ensure quality of life through the management of predictable complications. If abdominal distension does not improve in newborns who continue mechanical ventilation without lung problems, HD-related evaluation and surgery should be considered, and genetic testing should be done to check for Haddad syndrome.
Funding:This paper was supported by Fund of Biomedical Research Institute, Jeonbuk National University Hospital.
Conflict of Interest:No potential conflict of interest relevant to this article was reported.
Author Contributions:
Conceptualization: S.H.B., J.Y.J., K.J.K.
Supervision: J.Y.J.
Writing - original draft: J.S.Y., K.J.K.
Writing - review & editing: J.S.Y., S.H.B., J.Y.J., K.J.K.