Purpose This study aimed to evaluate postoperative outpatient follow-up practices among pediatric surgeons in Korea for five common congenital diseases: esophageal atresia with tracheoesophageal fistula (EA/TEF), anorectal malformation (ARM), Hirschsprung’s disease (HSCR), choledochal cyst (CC), and inguinal hernia (IH).
Methods A web-based survey consisting of 43 questions was distributed to members of the Korean Association of Pediatric Surgeons. The survey assessed the timing, frequency, and duration of outpatient follow-up, as well as disease-specific practices.
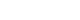
Results Of 154 invited surgeons, 45 (29.2%) responded. Most scheduled the first follow-up visit within one week after discharge. During the first postoperative year, follow-up visits were commonly held every three months, followed by six months or annual intervals. Most surgeons concluded follow-up before age 18; however, 15.6%–37.8% reported continuing follow-up into adulthood depending on the disease. Variation was observed in disease-specific practices: 44.4% routinely performed contrast studies for EA/TEF follow-up; sizes #14–15 Hegar dilators were most used in ARM; only 6.7% performed routine rectal irrigation in HSCR. For CC, 88.9% checked both blood tests and ultrasonography. Most IH patients received only one follow-up visit.
Conclusion While early postoperative follow-up practices among pediatric surgeons in Korea appear relatively consistent, wide variation exists in long-term strategies and disease-specific protocols. This reflects the tendency to rely on individual clinical judgment and highlights the need for standardized, national consensus.
Citations
Citations to this article as recorded by
Transition of Care to Late Adolescent/Adults in Paediatric Surgery: Perspectives From Indian Paediatric Surgeons Sunita Singh, Yogesh Kumar Sarin, Subhashis Saha, Mukesh Shukla Journal of Indian Association of Pediatric Surgeons.2026; 31(4): 541. CrossRef
Jinyoung Park, Dayoung Ko, Eun-jung Koo, Hyunhee Kwon, Ki Hoon Kim, Dae Yeon Kim, Seong Chul Kim, Soo-Hong Kim, Wontae Kim, HaeYoung Kim, Hyun-Young Kim, So Hyun Nam, Jung-Man Namgoong, Junbeom Park, Taejin Park, Min-Jung Bang, Jeong-Meen Seo, Ji-Young Sul, Joonhyuk Son, Joohyun Sim, Soo Min Ahn, Hee-Beom Yang, Jung-Tak Oh, Chaeyoun Oh, Joong Kee Youn, Sanghoon Lee, Ju Yeon Lee, Kyong Ihn, Hye Kyung Chang, Yeon Jun Jeong, Eunyoung Jung, Jae Hee Chung, Min Jeong Cho, Yun-Mee Choe, Seok Joo Han, In Geol Ho, Jeong Hong
Adv Pediatr Surg 2025;31(1):8-15. Published online May 28, 2025
Purpose This study aims to investigate and compare the incidence, demographic characteristics, clinical manifestations, preoperative diagnostic methods, anatomical classifications, associated anomalies, operative treatments, and postoperative outcomes of patients with intestinal atresia treated by the members of the Korean Association of Pediatric Surgeons (KAPS) through three nationwide surveys.
Methods KAPS conducted 3 national surveys in 1998, 2010, and 2024 to examine the patients diagnosed with intestinal atresia. In preparation for the survey, we developed a customized case registration form to obtain data on patient sex, birth weight, gestational age, clinical manifestations, preoperative diagnostic methods, anatomical types, associated anomalies, operative treatments, and postoperative outcomes. Authorized KAPS members completed the case registration form.
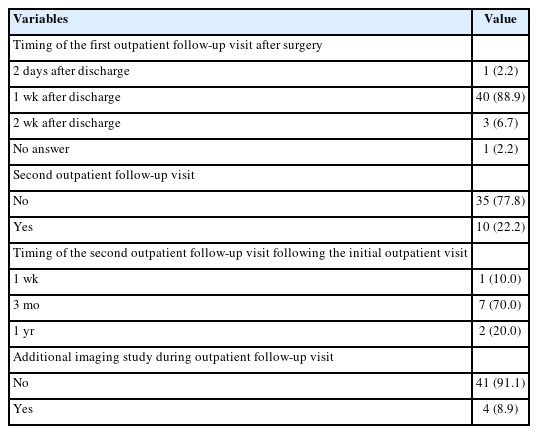
Results The first, second, and third national surveys included 218, 222, and 236 individuals diagnosed with intestinal atresia, respectively. The male-to-female ratios were 1.5:1, 1.1:1, and 1.1:1, respectively. The first, second, and third national surveys revealed that 34.3%, 43.3%, and 53.4% of patients were born before 37 weeks of gestation, respectively. Additionally, 28.7%, 32.0%, and 40.7% of patients had a birth weight under 2,500 g. In the third national survey, duodenoduodenostomy was the most common procedure, performed in 70 out of 82 patients diagnosed with duodenal atresia. Resection and anastomosis were the main surgical procedures conducted in 47 out of 54 cases of jejunal atresia and 74 out of 92 cases of ileal atresia. The mortality rates in the first, second, and third national surveys were 13.8%, 3.6%, and 1.3% respectively, with the lowest rate observed in the third national survey.
Conclusion These national surveys offer valuable insights into the current state of intestinal atresia, including specific surgical interventions and postoperative outcomes in South Korea. For pediatric surgeons aiming to enhance their understanding of intestinal atresia and its treatment options, these surveys could be an indispensable resource and guide.
Purpose The establishment of enteral feeding is the end point of any intestinal anastomosis. This study examined the effects of early feeding (EF) as compared to delayed feeding (DF) on postoperative outcomes after intestinal anastomosis in children.
Methods This was a randomized controlled pilot study to assess the effect of EF vs. DF in terms of time to reach full feed, along with wound infection and anastomotic leak.
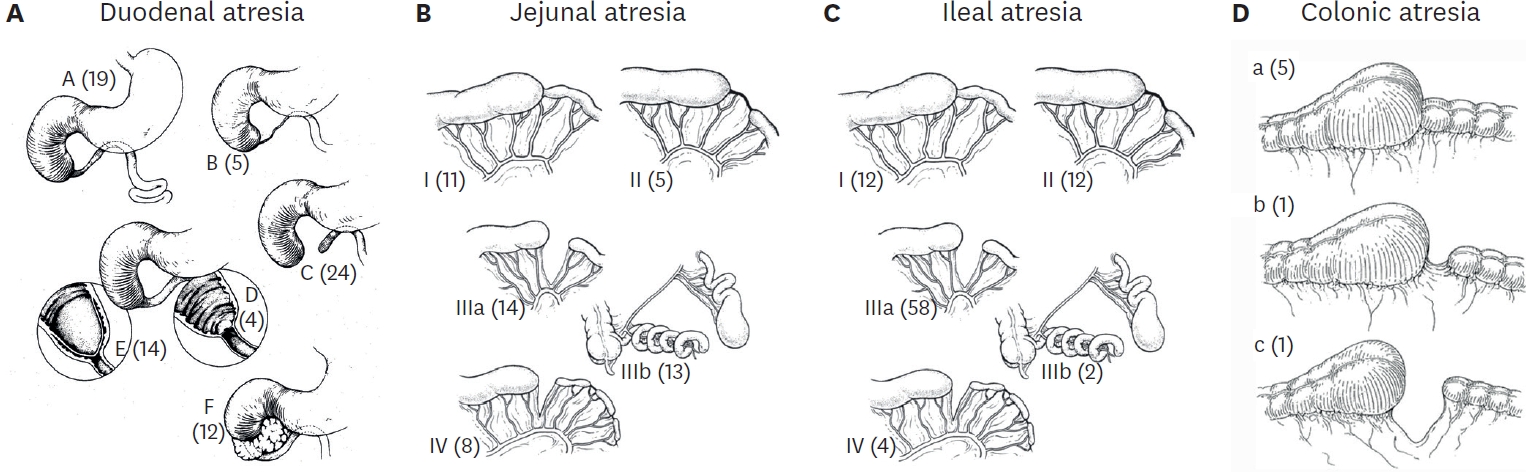
Results Twenty-eight patients were enrolled in both study groups. The median time to first feed in EF was 60 hours and 96 hours in DF. The median time to first bowel sound was 42 hours in EF and 48 hours in DF (p=0.208). The median time to first bowel movement was 72 hours in EF and 72 in DF (p=0.820). The median time of postoperative hospital stay was 5.5 days in EF and 6.0 days in DF (p=0.01). There was no significant difference in complications of wound infection, wound dehiscence, relook surgery, or anastomotic leak in both groups.
Conclusion EF after intestinal anastomosis is safe and feasible in children after intestinal anastomosis.
Citations
Citations to this article as recorded by
Modified Enhanced Recovery After Surgery Protocols in Pediatric Gastric Transposition: Effects on Recovery and Outcomes Mohit B. Chauhan, Nitin James Peters, Muneer Abas Malik, Shivani Dogra, Ravi Prakash Kanojia, Rajni Sharma, Sandhya Yaddanapudi, Monika Bawa, Shailesh Solanki, Jai Kumar Mahajan Journal of Indian Association of Pediatric Surgeons.2026; 31(1): 20. CrossRef
Early vs. delayed feeding after pediatric gastrointestinal surgery: a systematic review and meta-analysis Mohammed Al Blooshi Pediatric Surgery International.2026;[Epub] CrossRef
Nutritional Timing in Pediatric Stoma Reversal: Early Versus Late Feeding Practices Nadia Shoukat, Mumtaz Ahmed Qureshi, Ali Hasham, Sadia Shoukat, Nazia Azam Yousfani, . Matiullah Biological and Clinical Sciences Research Journal.2025; 6(9): 85. CrossRef
Intestinal failure (IF) is a term used to define the state where intestine’s function is significantly reduced, to the point where adequate growth and hydration cannot be maintained. In such cases, intravenous nutritional support is essential for sustaining the patient’s life. In pediatric patients, the most common cause of IF is short bowel syndrome (SBS). Due to the prolonged treatment and high complication rates, management of SBS remains a continuous challenge to many physicians. Herein, we report the case of a 2,260 g premature female infant born at 35-week gestational age with type 4 jejunoileal atresia. She presented with ultrashort bowel syndrome, having a bowel length of less than 15 cm, but ultimately achieved gut autonomy and restored bowel function through successful intestinal rehabilitation within the first two years of life.
A ciliated foregut cyst is a rare developmental anomaly. It develops from the primitive foregut. It is usually located supra-diaphragmatically. Its localization in the gallbladder is very infrequent and has been sparsely reported. We report a rare case of a ciliated cyst of the gallbladder in an 11-year-old female, who presented with complaints of upper abdominal pain for 2 months. She was suspected to have gallbladder duplication or gallbladder diverticulum on imaging. The histopathology reported this anomaly as a ciliated foregut cyst. The ciliated cyst of the gallbladder is a benign congenital lesion. Abdominal ultrasonogram and computed tomography/magnetic resonance imaging are suggestive of a cystic lesion of the gallbladder. The definitive diagnosis is by histopathological examination. This is a rare clinicopathological condition in the pediatric age group. The recommended treatment is laparoscopic cholecystectomy. The role of conservative management has not been established due to the rarity of the condition.
Proliferative myositis (PM) is a rare benign soft tissue neoplasm with a distinctive pseudosarcomatous proliferative reaction of muscles in tumors. Its rapid growth and bizarre microscopic appearance often require a differential diagnosis from a sarcomatous lesion. It has been reported occasionally, mostly as case reports in adult patients. Herein, we present a neonatal case of PM. To the best of our knowledge, this is the first report in the neonatal period.
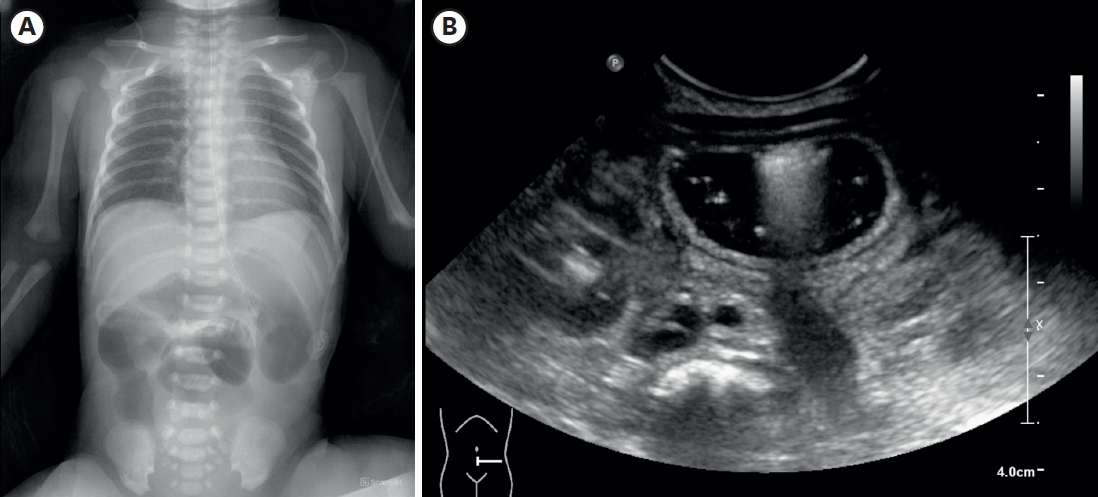
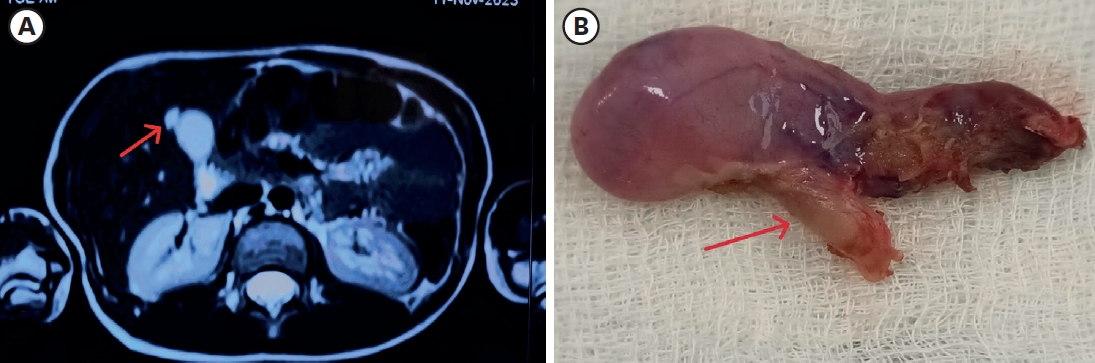
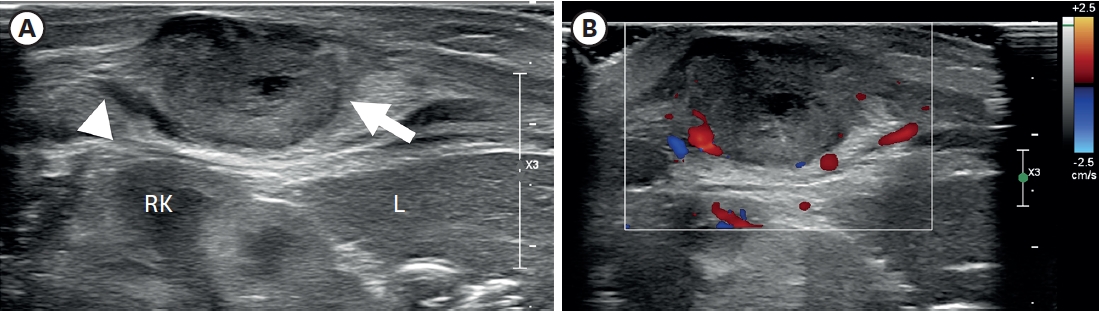
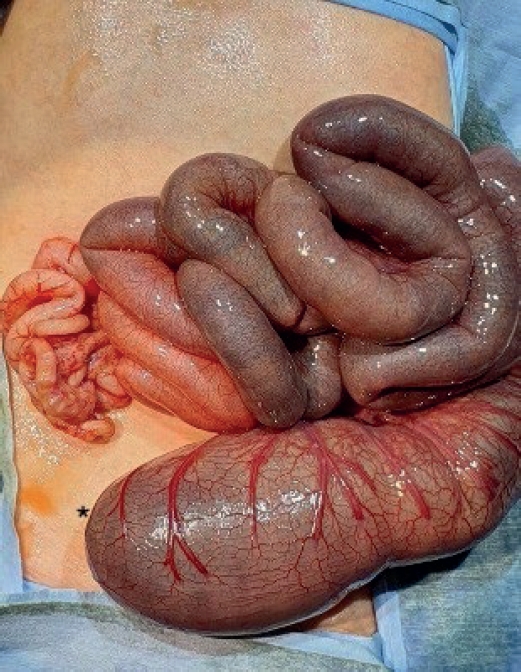
The concurrent occurrence of colonic atresia, malrotation, and Hirschsprung’s disease in neonates is extremely rare. These anomalies often share embryologic origins and present overlapping clinical symptoms that complicate diagnosis and management. We report two neonatal cases with this rare triad. Case 1 involved a term neonate initially diagnosed with esophageal atresia and later found to have colonic atresia, malrotation, and Hirschsprung’s disease. Case 2 was a preterm neonate presenting with abdominal distension and perforation, ultimately diagnosed with the same triad. Both underwent staged surgical management, including Duhamel’s procedures after confirming aganglionosis. Awareness of the possible coexistence of these anomalies is essential in neonates with colonic atresia and non-fixed colon. Surgical planning should anticipate aganglionosis and include rectal biopsy. This report emphasizes the importance of early suspicion and multidisciplinary approach for optimal outcomes.



 First
First Prev
Prev