Citations
Esophageal atresia (EA) is a diverse disease entity. We present a case of long gap EA without fistula corrected through totally laparoscopic and thoracoscopic esophageal replacement using gastric tube. A male baby weighing 3,000 g, with suspicion of EA, was born at gestational age of 37+6 weeks. Gastrostomy was made at an age of two days; seven months later, definite operation was planned. We determined to perform the gastric tube replacement due to long gap revealed by fluoroscopy. Gastric mobilization, gastric tube formation, and pyloroplasty were performed laparoscopically. An isoperistaltic 9 cm gastric tube was made using 2 Endo GIA 45, and interrupted end-to-end esophago-esophagostomy was performed thoracoscopically. With laparoscopy, gastropexy to the diaphragm was performed through the interrupted suture. Operation time was 370 minutes; there was no intraoperative event. Postoperative course was uneventful. He underwent esophageal balloon dilatation due to anastomosis stenosis in the months after surgery.
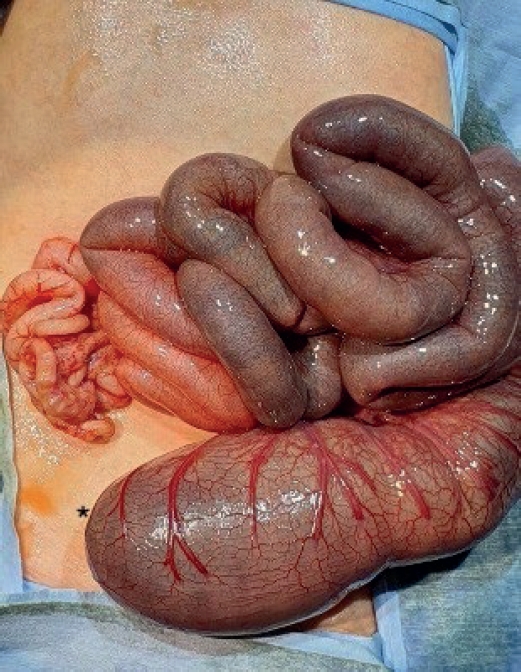
Santulli enterostomy has been used for various surgical abdominal conditions that require temporary diversion of bowel during a neonatal period. The aim of this study was to report clinical outcomes of Santulli enterostomy and to evaluate its usefulness.
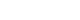
Between January 2000 and December 2016, 40 neonates who underwent Santulli enterostomy were enrolled; Santulli enterostomies were performed for 25 patients without previous laparotomy (primary Santulli group) and 15 patients with previous laparotomy (secondary Santulli group).
Small bowel atresia is the first common indication of Santulli enterostomy (22/40, 55.0%), and luminal discrepancy between proximal and distal bowel was the most common determinant factor of Santulli enterostomy (17/40, 42.5%). The median age at surgery and mean birth weight were 2 days and 2,480 g respectively in the primary group, and 71 days, 2,340 g respectively in the secondary group. Operation time was significantly longer in the secondary group than the primary group (156±48 minutes vs. 224±95 minutes, p=0.019), and there was no difference in the time taken to initiation of oral feeding between the two groups. Santulli enterostomy closure was performed at median 65 days after Santulli enterostomy for primary group and 70 days for secondary group. Six complications (15.0%) were found after Santulli enterostomy, and nine complications (24.3%) after Santulli enterostomy closure (p=0.302). The incidence of complications was significantly higher in secondary group than in primary group (4.5% vs. 53.3%, p=0.001), and the reoperation rate was also significantly higher in the secondary group (4.5% vs. 46.7%, p=0.004).
Santulli enterostomy could be applied as a temporary enterostomy in neonatal patients with various surgical abdominal diseases. Considering the high complication rate after secondary Santulli enterostomy closure, decision making on the timing of enterostomy closure should be done with caution.
Citations
A baby was diagnosed with esophageal atresia (EA) with tracheoesophageal fistula (TEF) on the next day after birth, and end-to-end anastomosis of esophagus with TEF ligation was performed. The distance between proximal and distal esophageal pouch was checked as 3 vertebral body lengths and a 1 cm-sized bronchogenic cyst (BC) was identified near carina on the right side, just below the proximal esophageal pouch. This case report described the baby who have a BC was located between the both esophageal pouch and a longer esophageal gap than usual EA with distal TEF.
Citations
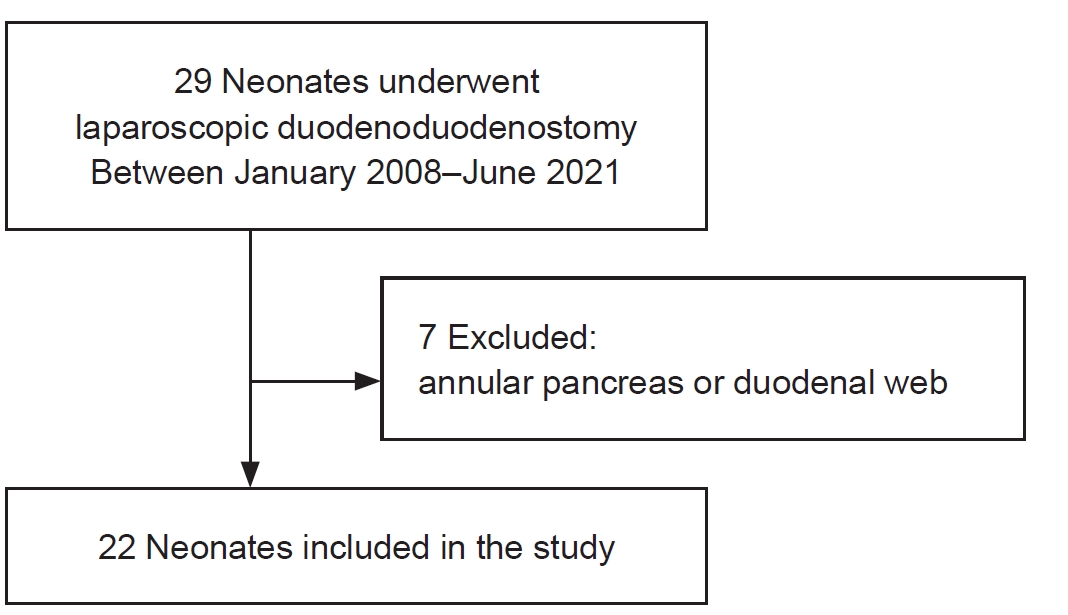
The Korean Association of Pediatric Surgeons (KAPS) performed the second nationwide survey on biliary atresia in 2011. It was a follow-up study to the first survey, which was performed in 2001 for the retrospective analysis of biliary atresia between 1980 and 2000. In the second survey, the authors reviewed and analyzed the clinical data of patients who were treated for biliary atresia by the members of KAPS from 2001 to 2010. A total of 459 patients were registered. Among them, 435 patients primarily underwent the Kasai operation. The mean age of patients who underwent the Kasai operation was 66.2±28.7 days, and 89.7% of those patients had type III biliary atresia. Only five patients (1.4%) had complications related to the Kasai operation. After the Kasai operation, 269 (61.8%) of the patients were re-admitted because of cholangitis (79.9%) and varices (20.4%). One hundred and fifty-nine (36.6%) of the patients who underwent the Kasai operation subsequently underwent liver transplantation. The most common cause of subsequent liver transplantation was persistent hyperbilirubinemia. The mean interval between the Kasai operation and liver transplantation was 1.1±1.3 years. Overall the 10-year survival rate after the Kasai operation was 92.9% and the 10-year native liver survival rate was 59.8%. We had 23 patients for primary liver transplantation without the Kasai operation. The mean age patients who underwent primary liver transplantation was 8.6±2.9 months. In summary, among the 458 Kasai-operation and liver-transplantation patients, 373 lived, 31 died, and 54 were unavailable for follow up. One-third of the patient who survived have had complications correlated with biliary atresia. In comparison with the first survey, this study showed a higher survival rate and a greater number of liver transplantation.
Citations
Colonic atresia (CA) is the rare cause of intestinal obstruction, and diagnosis of CA is difficult. But only few research has been performed, so little information has been available. The purposes of this study was to analyze the clinical findings of CA so that help physicians make decision properly. Children with CA who were treated at the division of pediatric surgery at Asan Medical Center in the period from January 1989 to December 2011 were evaluated retrospectively. A total of 6 children were treated with CA. These accounted for 2.7% of all gastrointestinal atresias managed in Asan Medical Center. Only one child was premature and low birth weight, the others were fullterm neonates and showed normal birth weight. Vomiting and abdominal distension were common symptoms and simple X-ray and barium study were used for diagnose of CA. But only 66.7% of the babies were diagnosed as CA pre-operatively. And 2 children out of 6 underwent re-operation due to missed CA at the time of the first operation. In aspect of types of atresia, the type IIIa were two, type IV were two, type I was one case, and one child showed rectal stenosis due to rectal web. Various operations were done according to individual findings and associated diseases. The 50% (n=3) of children underwent the primary anastomosis and the others (n=3) underwent colostomy first and staged operation later for missed CA or associated disease. All of them were recovered any significant complications. Therefore, the prognosis of CA is satisfactory if diagnosis and surgical management could be made properly. But because of the low incidence of CA, delay of diagnosis and treatment may occur. To prevent delay of diagnosis, we suggest prompt evaluation of doubtful infant and careful inspection of distal patency of bowel including whole colon and rectum when operating patients with intestinal atresia at any level.
The purpose of this study is to analyse clinical impact of specific MRI findings in liver in patients of long-term survivors after Kasai portoenterostomy (KPE). Twenty-eight patients who were underwent KPE were followed up more than 5 years. Macro-regenerative nodule (MRN) and beaded-duct dilatation (BDD) were considered as important findings in liver MRI. The association between these findings in MRI and clinical indicator, serum bilirubin level and history of cholangitis were evaluated. Sixteen patients (57.1%) were shown MRN in liver MRI. There were 14 patients(50%) whose MRI showed BDD. Serum total and direct bilirubin were 3.6mg/dL and 1.8mg/dL respectively in positive MRN group whereas 1.4mg/dL and 0.7mg/dL in negative MRN group (
Hiatal hernia is a very rare disease in the pediatric population. However information from our esophageal atresia postoperative follow-up program has hypotheses; “Hiatal hernia may more frequently occur in postoperative esophageal atresia patients (EA group) than in the general pediatric population (GP group)” and “The tension on the esophagus after esophageal anastomosis may be an important etiologic factor of hiatal hernia in EA group”. To prove the first hypotheses, we compared the incidence of hiatal hernia in the GP group with the incidence in the EA group. The Incidence in the GP group was obtained from national statistic data from Statistics Korea and Health Insurance Review and Assessment Service of Korea. The incidence in the EA group was obtained from the medical record and the imaging studies of our esophageal atresia postoperative follow-up program. To prove the second hypothesis, the presumptive risk factors for the development of hiatal hernia in EA group, such as the type of esophageal atresia, degree of esophageal gap, the stage operation and the redo-operation with resection and re-anastomosis of esophagus were analyzed statistically. The total number of patients in the EA group was ninety-nine and there were 5 hiatus hernias. The incidence of EA group (5 %) is significantly higher than incidence of GP group (0.024 %). (
Citations
Tracheoesophageal fistula without esophageal atresia (H-type TEF) is a congenital anomaly that is characterized by a fistula between the posterior wall of the trachea and the anterior wall of the esophagus, not accompanied by esophageal atresia. The purpose of this study is to investigate the clinical characteristics, diagnostic time, the side of cervical approach and short term result after surgery by searching medical records of patients treated for H-type TEF. The search was done at University of Ulsan, Department of Pediatric Surgery of Asan Medical Center, and the total number of patients from May 1989 to December 2010 was 9 with M:F ratio of 1:2. The median gestational age was 39(+6) (32(+6)~41(+0)) wks. Seven out of nine patients were born at term and the other two were born premature. The clinical presentation was aspiration pneumonia, difficulty in feeding, chronic cough, vomiting, abdominal distension and growth retardation. The symptoms presented right after birth. The diagnosis was made with esophagography and the median time of diagnosis was 52 days of life. The majority of surgical corrections were performed within two weeks of diagnosis (median; 15d, range; 1d - 6m). Six patients had associated anomalies, and cardiac anomalies were most common. The cervical approach was utilized in all cases (right 2, left 7). Transient vocal cord palsy and minor esophageal leakage complicated two cases. Although the diagnosis of H-type TEF was difficult and often delayed, we had a good short term result. The left cervical approach was preferred.
Citations
The onset of hypertrophic pyloric stenosis in the postoperative course of esophageal atresia with tracheoesophageal fistula is rarely reported. The diagnosis could be delayed due to its mimicking symptoms of other postoperative complications including gastroesophageal reflux or anastomotic stricture. We present an infant who had surgery for esophageal atresia with tracheoesophageal fistula. He had never fed since birth. The infant presented with an increased amount of orogastric tube drainage and consistently distended gastric air on simple abdominal X-ray. Abdominal ultrasonography showed hypertrophic thick pyloric muscle. The diagnosis of pyloric stenosis was confirmed d is rarely reported. The diagnosis could be delayed due to its mimicking symptoms of other postoperative complications including gastroesophageal reflux or anastomotic stricture. We present an infant who had surgery for esophageal atresia with tracheoesophageal fistula. He had never fed. The infant presented with uring surgery. After pyloromyotomy, the patient's condition improved.
Citations
This study was aimed to evaluate associated congenital anomalies in the patients with esophageal atresia with tracheoesophageal fistula (EA/TEF). Forty-two neonates with the diagnosis of EA/TEF treated over a 10 year period in a single institution were included in this study. The demography of EA/TEF was analyzed. Major associated anomalies including vertebral, anal, cardiac, renal, limb, neurologic and chromosome were reviewed and categorized. Males were slightly more dominant than females (1.47:1) and all patients had Gross type C EA/TEF. Only 19% of the patients had solitary EA/TEF without associated anomalies. Cardiac anomalies were the most common associated congenital anomaly in patients with EA/TEF (73.8%). But 47.6% were cured spontaneously or did not affect patients' life. Atrial septal defect (ASD) was the most common cardiac anomaly followed by patent ductus arteriosus (PDA) and ventricular septal defect (VSD). Among gastrointestinal anomalies (23.8%), anorectal malformations were the most frequent, 70% Vertebral and limb abnormalities accounted for 11.9% and urogenital malformations 9.5% of the anomalies in patients with EA/TEF. VACTERL associated anomalies were 23.8% and 4.8% had full VACTERL. Almost 12% of EA/TEF had neurologic anomalies. Patients with EA/TEF require preoperative evaluation including neurologic evaluation to detect anomalies not related to VACTERL. Though associated cardiac anomaly occurred in 73.8% of patients in our study, only 21.42% needed surgical correction. The authors suggesrs further studies with large numbers of patients with EA/TEF.
Citations
Biliary atresia (BA) is an infantile cholestatic disease of progressive obliterative cholangiopathy with varying degrees of damage to both extra and intrahepatic bile ducts due to unknown causes. The diagnostic studies should be done to diagnose or exclude BA without unnecessary delay. Kasai portoenterostomy is the first choice of treatment for bile drainage from microscopic bile ductules present in the portal fibrous mass. The medical management after Kasai portoenterostomy should be done carefully to maintain bile excretion and prevent and treat complications including cholangitis, hepatic fibrosis, portal hypertension and nutritional problem. The reported five years-survival rates after Kasai portoenterostomy range from 30 to 60%. About 20% of all patients undergoing Kasai portoenterostomy during infancy survive into adulthood with their native liver. Even if Kasai portoenterostomy remains as the first line of treatment in BA, liver transplantation serves as a good salvage treatment when portoenterostomy fails or liver function gradually deteriorates after initially successful establishment of bile flow. Overall 5-year survival rate in BA is about 90% in recent series.
Tracheal injury is a rare complication of endo-tracheal intubation. However in neonates, the rates of morbidity and mortality are high. Recommendations for treatment are based on the several reports of this injury and are individualized. Conservative management can be effective in some cases. We describe the case of a neonate who presented with subcutaneous emphysema after intubation in a neonatal intensive care unit. This patient suffered full VACTERL syndrome and had 1.7mm diameter subglottic stenosis. Conservative management resulted in no further increase in subcutaneous emphysema and after 10 days the patient was stable.
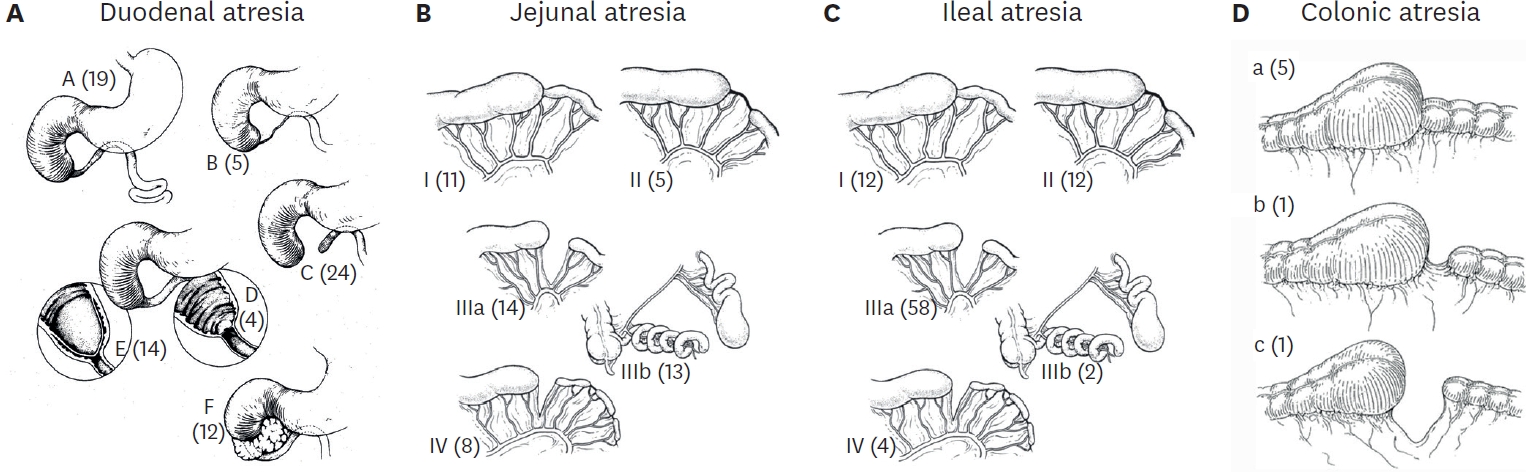
The members of the Korean Association of Pediatric Surgeons conducted a retrospective study of two hundred and twenty-two cases of intestinal atresia for the period from January 1, 2007 to December 31, 2009. Seventeen hospitals were involved. There were 76 duodenal, 65 jejunal, and 81 ileal atresias (3 colonic). The male to female ratio was 0.85:1 in DA and 1.34:1 in JIA. Ninety-four patients(43.3%) were premature babies (DA 40.3%, JA 64.6%, IA 28.8%), and 70 babies (32.0%) had low birth weight (DA 38.7%, JA 44.4%, IA 16.0%). Antenatal diagnosis was made in 153 cases (68.9%). However, 27 infants (17.6%) with antenatal diagnosis were transferred to the pediatric surgeon's hospitals after delivery. Maternal polyhydramnios was observed in 81 cases (36.59%) and most frequent with proximal obstruction. In forty-four cases (19.8%), only simple abdominal film was taken for diagnostic study. The associated malformations were more frequently observed in DA - 61.8% in DA and 22.6% in JIA. Meconium peritonitis, small bowel volvulus and intussusception were more frequently associated with ileal atresia. The overall mortality rate was 3.6%.
Citations
Although the incidence of esophageal atresia (EA) is higher in twins than in singletons by two to three times, EA usually affects only one member of twins. We report one pair of twins concordant for EA. A 31-year-old healthy woman bore monozygotic female twins at 36 weeks of gestation. They weighed 2,216 and 2,480g, respectively. They had EA with distal tracheoesophageal fistula and underwent primary esophageal anastomosis on the birth day and the 2nd day of life, respectively. Twin A also had suspicious antral obstruction and pyloroplasty was done simultaneously with esophageal repair. She needed antral web excision for continued gastric stasis one month after 1st operation and three balloon dilatations of the esophagus. Twin B recovered uneventfully.
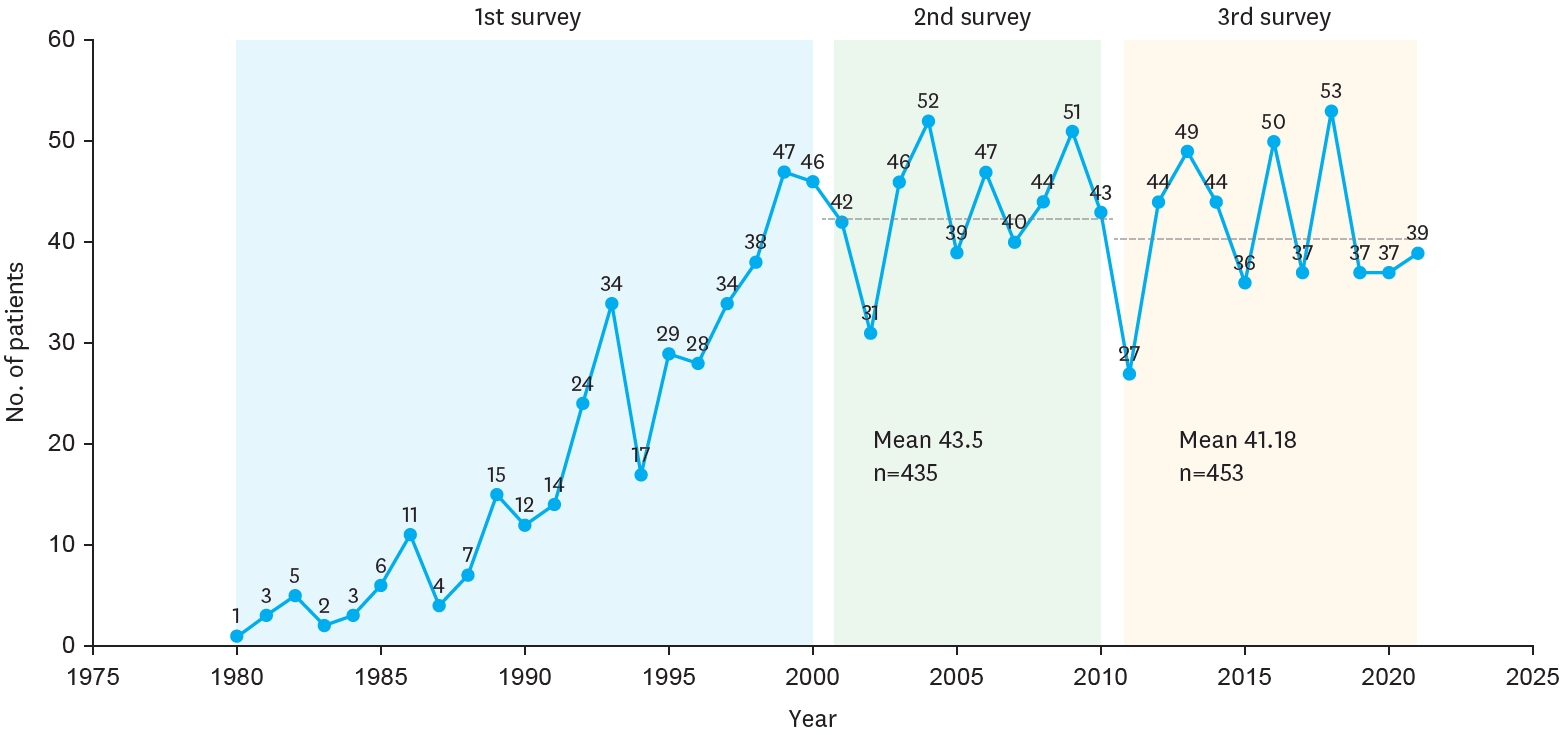
Situs inversus abdominis is a rare congenital condition commonly associated with serious cardiac and splenic malformations. The importance of recognizing the presence of situs inversus abdominis preoperatively is emphasized by the fact that the surgical incision is placed on the incorrect side of the abdomen. A 6 day-old girl was referred to our hospital because of bile stained vomiting. A plain radiography of abdomen and chest showed the heart to be normal position and a reversed "double-bubble" picture with no other gas shadow in the rest of the abdomen. Abdominal computed tomography scan revealed situs inversus with the stomach and polysplenia on the right side and the liver on the left side. A laparotomy confirmed the diagnosis of situs inversus with duodenal atresia. The obstruction was bypassed by constructing a side-to-side duodenoduodenostomy. The postoperative course was uneventful.
Citations
To evaluate the long-term prognosis of biliary atresia after Kasai operation, a total of 14 patients (of the 41 patients operated upon from 1982 to 1997), who had been followed up for more than 10 years, were included in this retrospective study. Eleven out of 14 patients survived with their native livers, and their data analyzed for age at operation, clearing time of jaundice, histological outcome, postoperative complications, effectiveness after the application of an intussusception anti-reflex valve, and quality of life. Average age at surgery was 62.8 days. Serum bilirubin was normalized within three months in all patients. Six among the eleven long-term survivors had ascending cholangitis as one of the postoperative complications. The application of an intussusception anti-reflux valve did not show any statistical significance in long-term survival. Most of long-term survivors appeared to enjoy good quality of life. Kasai operation might not be the definitive treatment for biliary atresia; however, Kasai operation made it possible to achieve long-term survival for patients with biliary atresia when the patients were detected and treated as early as possible.
Children who underwent reparative operations for esophageal atresia (EA) with or without tracheoesophageal fistula (TEF), are confronted with many gastrointestinal or respiratory problems, especially during the early years of life. We reviewed the medical records of 50 patients who underwent repairs of EA with or without TEF at the Division of Pediatric Surgery, Samsung Medical Center, from December 1994 to December 2005. Current status of children was accessed by telephone-interview, but only 27 of them were accessible. Of 50 patients, 3 patients (6%) were type A, 45 patients (90%) were type C, and 2 patients (4%) were type E. The mean interval between primary operation and interview was 5.5 years. The incidences of growth retardation (<10 percentile of height/weight) were 39% and 21 % during the first 5 years after repairs, respectively. The incidences of dysphagia or gastroesophageal reflux and recurrent respiratory infections were 33% and 39 %, respectively. However, these problems were likely to improve as the children grew. The incidences of growth retardations (<10 percentile of height/weight) were 11% and 11% for the children more than five years postoperative. The incidences of dysphagia or gastroesophageal reflux and recurrent respiratory infections were 22% and 22%, respectively. Children with EA with or without TEF are faced with many obstacles. Close observation and adequate treatment for delayed postoperative complications are necessary to improve the quality of life for these children.
Citations
Esophageal atresia with double tracheoesophageal fistula is a very rare anomaly and is difficulty to diagnose preoperatively. We treated a full term baby with esophageal atresia with double tracheoesophageal fistula. At the first operation, only the distal tracheoesophageal fistula was identified and ligated. When the upper esophageal pouch was opened, intermittent air leakages in sequence with positive bagging were noticed. However, intraoperative bronchoscopy did not identify a fistula in the proximal pouch, and the operation was completed with end to end anastomosis of the esophagus. On the 7th postoperative day, esophagography showed another tracheoesophageal fistula proximal to the esophageal anastomosis. A wire was placed in the fistula preoperatively under bronchoscopy. At the 2nd operation through the same thoracotomy incision the proximal fistula was identified and ligated. On the 12th postoperative day, esophagography showed neither stricture nor leakage.
There is significant morbidity and mortality associated with the combination of esophageal atresia (EA) and duodenal atresia (DA). Nevertheless, the management protocol for the combined anomalies is not well defined. The aim of this study is to review our experience with the combined anomalies of EA and DA. From May 1989 to August 2006, seven neonates were diagnosed as EA with DA at Asan Medical Center. In all cases, the type of EA was proximal EA and distal tracheoesophageal fistula (TEF). The diagnosis of DA was made in theprenatal period in 1, at birth in 4, 4 days after birth in 1 (2 days after EA repair) and at postmortem autopsy in 1. Except the one case where DA was missed initially, primary simultaneous repair was attempted. DA repair with gastrostomy followed by EA repair in 2, EA repair followed by DA repair without gastrostomy in 2, and TEF ligation followed by DA repair with gastrostomy in 1. There were two deaths. One baby had a large posterolateral diaphragmatic hernia, and operative repair was not attempted. The other infant who had a TEF ligation and DA repair with gastrostomy expired from cardiac failure due to a large patent ductus arteriosus. Simultaneous repair of EA and DA appears to be an acceptable management approach for the combined anomalies, but more experience would be required for the selection of the primary repair of both anomalies.
Citations
From 1979 to 2006, fifty eight patients with esophageal atresia were treated by one pediatric surgeon at Hanyang University Hospital. We analyzed the clinical findings and outcome of these 58 patients. There were 30 males and 28 females. Their mean birth weight was 2,960 ± 400 g (1,170~4,020 g). The most common type of anomaly was Gross type C (49 patients; 84.5 %). There was no type B. Fifty-two patients underwent definitive surgery. Postoperative complications were as follows: anastomotic leakage in 17 patients (32.7 %), anastomotic site stricture in 15 (28.8 %), gastroesophageal reflux in 10 (19.2 %) and recurrent TEF in 1 (1.9 %). A total of 152 associated anomalies were detected from 48 patients (82.2 %). The cardiovascular system was the most commonly affected (30 patients with 46 anomalies). The VACTERL association was present in 14 patients (24.1 %). Operative mortality was 17.3 % including self-discharge patients after operation.
Citations
The aim of this study is to analyze the outcomes of the esophageal atresia with tracheoesophageal fistula over the last 2 decades. The records of 51 patients born between 1987 and 2006 were reviewed. Twenty-seven patients were male. Mean values of the age, gestational age and birth weight were 2.9 days, 296 days and 2.7kg, respectively. All patients had Gross type C anomalies. Thirty-one patients (60.7 %) had one or more associated congenital anomalies and the most common anomaly was cardiac malformation. In 48 cases, primary anastomosis was done and staged operation was done in one case. Circular myotomies in the proximal esophagus were performed in 9 cases. Postoperative complication developed in 26 cases (54 %): pulmonary complication in 12 cases, anastomotic leakage in 10 and anastomotic stricture in 10, recurrent trachoesophageal fistula in one and tracheomalasia in 2 cases. Reoperation was carried out in 2 patients with anastomotic leaks, the remaining leaks were managed non operatively. Three of the strictures were reoperated upon and the others were successfully managed by balloon dilatations. Overall mortality rate was 15.6 %. Mortality rate of the second 10 years (8 %) period decreased significantly compared to that of the first 10 years (23 %) period.
Citations
Biliary atresia (BA) is an uncommon neonatal surgical disease that has a fatal outcome if not properly treated. The survival rates of the patients with native liver after Kasai's operation in countries outside Japan are not so good. We reviewed the results of 22 cases of biliary atresia treated in Kosin University Hospital between October 1987 and March 2001. There were 13 males and 9 females aged from 21 to 106 days (mean 52 days). There were 3 cases of Type I (13.6%), and 3 of Type II (13.6%), and 16 Type III (72.7%). The operative methods were resection of the common bile duct remnant and cyst followed by Roux-en-Y hepaticojejunostomy in 3 cases for Type I BA; Kasai I in 15 cases, Kasai II in 1 case, and Ueda's operation in 3 cases for Types II and III BA. There was no death within the first 30 days after operation. We were able to follow 21 of the 22 patients (95.4%) for more than 5 years. The actual 5 year survival rate (YSR) was 40.9%. One Type I case received a living-related liver transplantation at 6 years of age because of the multiple intrahepatic stones and liver cirrhosis. Five YSR after biliostomy group (Kasai II and Ueda op.) was 75% (3/4) while that of Kasai I was 20% (3/15). One case had no bile duct in the resected fibrotic plaque on microscopic review and died 8 months after Kasai I operation, would have been a strong candidate for early liver transplantation. From the above result, our conclusions are as follows; (1) early liver transplantation should be considered for cases of no bile duct after pathologic examination of the resected specimen, (2) measures to prevent postoperative cholangitis and prevention of postoperative liver cirrhosis are needed, (3) liver transplantation program should be available for failed cases.
We reviewed the records of 25 patients who were re-operated upon after primary repair of esophageal atresia with or without fistula at the Department of Pediatric Surgery, Seoul National University Children's Hospital, from January 1997 to March 2007. Types of the esophageal atresia anomalies were Gross type A in 5 patients, C in 18, and E in 2. The indications for re-operation were anastomosis stricture (n = 14), tracheo-bronchial remnant (n = 4), persistent anastomosis leakage (n = 3), recurrent tracheo-esophageal fistula (n = 2) and esophageal web (n = 2). The interval between primary and secondary surgery was from 48 days to 26 years 5 months (mean: 2 years and 4 months). Four patients required a third operation. The interval between the second and third operation was between 1 year 1 month and 3 year 10 month (mean: 2 years 5 months). Mean follow up period after last operation was 35 months (1 years–8 years 6 months). The secondary surgery was end-to-end esophageal anastomosis in 15, esophagoplasty in 5, gastric tube replacement in 5. After secondary operation, 6 patients had anastomosis stricture (4 patients were relieved of the symptoms by balloon dilatation, 2 patients underwent tertiary operation). Five patients had leakage (sealed on conservative management in all). Two patients had recurrent tracheo-esophagel fistula (1 patient received chemical cauterization and 1 patient underwent tertiary operation). Currently, only one patient has feeding problems. There were no mortalities. Secondary esophageal surgery after primary surgery for esophageal atresia was effective and safe, should be positively considered when complications do not respond to nonoperative therapy.
Tapering enteroplasty was first described by Thomas in 1969 as one method of intestinal anastomosis. The advantages of tapering enteroplasty in the intestinal atresia are: First, it makes end-to-end anastomosis possible between the atretic bowel ends with considerable differences in diameters. Second, it promotes the recover of the postoperative bowel function. Third, it prevents the possibility of the short bowel syndrome by eliminating the need of resecting the dilated bowel. A total of 22 patients with intestinal atresia who underwent tapering enteroplasty from January 1988 to December 2005 at our institute were reviewed. In 3 of 22 cases, tapering enteroplasty was the 2nd operation after an initial end-to-oblique anastomosis. We reviewed the following items: age, sex, type and location of intestinal atresia, initial feeding and total enteral feeding start day, the length of hospital stay and complications. The average age of the patients was 7 days. Male to female ratio was 1 to 1.2 (10 cases: 12 cases). We performed the tapering enteroplasty on all types and locations of the intestinal atresia from the duodenum to the colon: type I (n=3), type II (n=4), type IIIA (n=7), type IIIB (n=5), type IIIB and IV (n=1), type IV (n=1) and type C (duodenum) and type IIIB and IV (jejunum). On the average, the oral feeds were started on the postoperative 8.8th day, and full caloric intake via the enteric route was achieved on postoperative 13.3th day. The average length of hospital stay was 19.6 days. There were 1 case (4.5 %) of anastomotic complication and 2 cases (9 %) of adhesive ileus among 22 patients. The tapering enteroplasty on all types of intestinal atresia is a usefull operative method when there are considerable diameter differences between the atretic bowel ends.
Citations
Biliary atresia (BA) is the result of fibrosing destructive inflammatory process affecting intrahepatic and extrahepatic bile ducts, which lead to cirrhosis and portal hypertension. Kasai portoenterostomy has been the standard operative procedure in biliary atresia. Recently, there has been remarkable increase in the survival rate in cases of BA. However, long-term survivors are not clearly evaluated in Korea. To define long-term prognosis factors of patients who underwent surgery for BA, a retrospective study was undertaken of 10 (37 %) patients surviving more than 10 years among 27 patients who underwent one of Kasai procedures between 1981 and 1995. Hepatomegaly was present in 4 and splenomegaly in 7 patients. Serum bilirubin was normalized at 1 year after operation. Aspartate aminotransferase (AST, GOT), Alanine aminotransferase(ALT, GPT) were normalized at 12 years and alkaline phosphatase(ALP) was normalized at 13.5 years. Cholangitis developed mainly within 5 years after operation so close follow up is needed. Life long follow-up is needed because of progressive deterioration of liver function even after 10 years.
The prognostic factors for extrahepatic biliary atresia (EHBA) after Kasai portoenterostomy include the patient's age at portoenterostomy (age), size of bile duct in theporta hepatis (size), clearance of jaundice after operation (clearance) and the surgeon's experience. The aim of this study is to examine the most significant prognostic factor of EHBA after Kasai portoenterostomy. This retrospective study was done in 51 cases of EHBA that received Kasai portoenterostomy by one pediatric surgeon. For the statistical analysis, Kaplan-Meier method, Logrank test and Cox regression test were used. A
Citations
VATER association is defined as a combination of 3 or more anomalies-vertebra(V), imperforate anus (A), esophageal atresia with or without tracheoesophageal fistula (TE), renal and radial anomaly(R). We reviewed our experiences in one center to determine etiology, prevalence, clinical manifestation, other associated anomaly and prognosis. Two hundred and twenty-three cases that underwent operations for imperforate anus or esophageal atresia were analyzed retrospectively through medical records at Department of Pediatric Surgery, Asan Medical Center from June, 1989 to July, 2005. The total number of neonates who had been admitted during period of study were 46,773 and VATER association was 9 (0.019 %, 1.92 persons per 10,000 neonates). Median gestational age and birth weight were 37+4wk (35+1 – 41+4) and 2,594 g (1,671–3,660), respectively and median age of mother was 32 years (23–38). There was no family history. Three patients were twins but their counterparts had no anomalies. Patients who have 3 anomalies were 6, 4 anomalies in two and 5 anomalies in one patient. Vertebra anomalies were detected in 7(77.7 %), imperforate anus in 8(88.9 %), esophageal atresia in 5 patients (55.6 %), renal anomaly in 6(66.7 %), and radial anomaly in 5(55.6 %), respectively. Four patients are alive, 2 patients were lost during follow up period. Three patients died due to neonatal sepsis, respiratory dysfunction and cardiac failure. VATER association did not appear to be a definite risk factor, but merely a randomized combination of 5 anomalies. The prognosis was dependent on the other associated anomalies, appropriateness of management and operation. Careful follow-up and aggressive treatmentare required for improving survival and quality of life.
The repair of esophageal atresia with a long gap still continues to pose difficulties for the surgeon. There is general agreement that the child's own esophagus is best, but it is also believed that a primary repair is not always possible. Foker JE et al. (1997) developed a technique of esophageal lengthening using external traction sutures. We experienced one case of esophageal atresia with a 4.5cm gap (4 vertebral spaces) which was repaired using the external traction suture technique.
Citations
Esophageal atresia without tracheoesophageal fistula accounts for 7-11% of all types of esophageal atresia and is very difficult to treat. In our hospital from 1990 to 2005, we operated upon 40 patients with esophageal atresia, and 6 had pure atresia. The preoperative characteristics, operative findings and post operative course of the six patients with pure atresia were analysed. Immediate gastrostomy was performed in all 6 patients. One patient had simultaneous cervical esophagostomy. Esophageal reconstruction procedures were transhiatal gastric pull up in 3 patients, esophagocologastrostomy utilizing left colon in 1, and transthoracic esophagoseophagostomy with esophageal bougination in 2. Postoperative complications were pneumonia, anastomosis leakage, and gastroesophageal reflux symptom. Conservative management was effective in all patients. A larger series of cases would be required to demonstrate the most effective treatment for this particular anomalous condition.
Citations
Intestinal atresia is a frequent cause of intestinal obstruction in the newborn. We reviewed the clinical presentation, associated anomalies, types of atresias, operative managements, and early postoperative complications in 36 cases of intestinal atresia treated at the Department of Surgery, Kyungpook National University Hospital between January 1994 and February 2003. Location of the lesion was duodenum in 17 patients, jejunum in 11 patients and ileum in 8 patients. The male to female ratio was 1:1.4 in duodenal atresia (DA), 2.7:1 in jejunal atresia (JA) and 7:1 in ileal atresia (IA). The most common type was type III (41.1 %) in DA, and type I (52.6 %) in JA and IA. The most common presenting symptoms was vomiting(88.2 %) in DA, but in jejunoileal atresia, vomiting(89.4 %) and abdominal distension(89.4 %) were the most common sign and symptom. All cases of DA were diagnosed by plain abdominal radiography. There were 6 cases of DA with congenital heart disease, 3 cases of DA with Down syndrome and 3 cases of JA with meconium peritonitis. Segmental resection was performed in 13 cases, duodenoduodenostomy in 11 cases, membrane excision in 7 cases, jejunojejunostomy in 2 cases, gastroduodenostomy in 2 cases and ileocolic anastomosis in 1 case. There were 9 postoperative complications including 3 each of anastomotic leakage, wound infection, and intestinal obstruction 3 cases. The mortality rate for DA was 11.8 %(2/17). Both deaths in DA were attributed to congenital heart disease. The mortality rate for JA was 18% (2/11). Both cases died with sepsis and short bowel syndrome.
Citations
The history of esophageal replacement in infants or children is the history of development of various kinds of alternative conduits such as stomach, colon, and small bowel. The gastric tube has been the most widely used conduit. From January 1988 to May 2003, 23 esophageal replacements with gastric tube were performed at the Department of Pediatric Surgery, Seoul National University Childrens Hospital. Statistical analysis was performed using Windows SPSS11.0 Pearson exact test. There were Gross type A(n=10), type B(n=1), type C(n=11), type D(n=1). Ten patients who had long gap esophageal atresia (type A-8, type B-1, type C-1) and 13 patients (type A 2, type C-10, type D-1) who had stenosis, leakage, recurred tracheoesophageal fistula, and esophagocutaneous fistula after previous corrective operations, had esophageal replacement with gastric tube. Mean follow-up periods were 4 year 2 months (7 months-15 year 1 month). There were postoperative complications including GERD in 16 (69.6 %), leakages in 7 (30.4 %), diverticulum at anastomosis in 2 (8.7 %), anastomosis site stenosis in 4 (17.3 %), and distal stenosis of the gastric tube in 1 (4.3 %). There was no statistical significance between operation types and postoperative leakage and gastroesophageal reflux. In conclusion, esophageal replacement with gastric tube may be a useful surgical option in esophageal atresia with long gap and esophageal atresia complicated by previous corrective operation.
Biliary atresia (BA) with extrahepatic biliary cysts (EHBC) is a rare disease. It has been generally recognized as type I (correctable with cystic dilatation), which means a good prognosis. From a total of 73 patients with BA who underwent operation from September 1988 to September 2003 at our institute, 7 (9.6 %) cases of type III BA with EHBC (uncorrectable with cystic dilatation) are reviewed. Clinical findings, laboratory data, radiologic findings, treatment methods and outcomes were reviewed. Female was more prevalent (male to female ratio; 2:5). All cases were type III with EHBC according to the intraoperative cholangiography, and underwent Kasai's portoenterostomy. The mean age was 57 days at operation. Three patients(42.9 %) are long term survivors. Further evaluation is needed to determine the correlation between prognostic factors and outcome for.
No abstract available.
This study reviews 14 years' experience of esophageal atresia with special emphasis on the clinical profile and the outcome. From May 1989 to February 2003, 65 cases of esophageal atresia (EA) were treated at Asan Medical Center. Boys outnumbered girls 2.4 to 1. Prematutity and low birth weight were 27.7% and 38.5%. Esophageal atresia with distal tracheoesophageal fistula (TEF) was the most common type (87.7%), followed by pure EA and H type fistula. Forty-six patients (70.8%) had one or more associated anomalies, cardiac malformations were the most common. Duodenal atresia was found in 7 cases. There were 6 patients (9.2%) with VATER cluster. VACTERL cluster was present in 18 patients (27.7%), one of who fulfilled the complete syndrome. Waterston group A, B and C made up 21.5%, 40.0% and 38.5% of the total group. Surgical treatment was attempted in 63 patients and deferred in 2 who had severe associated malformations. For EA with distal TEF, primary esophago-esophagostomy was carried out in 51 cases, and division of TEF and gastrostomy in 4 cases and no operation in 2 cases. For pure EA, colonic graft was done in 2 after gastrostomy and esophagostomy, and esophago-esophagostomy was performed in 2 after gastrostomy. Two TEF was carried out in 2 cases with H type TEF. The overall survival rate was 76.9%, and survival by Waterstuon classification was 100% in group A, 80.8% in B and 60.0% in C. Thorough workup for associated anomalies, interdepartmental approach and more careful surgical decision and technique are required to improve the outcome of EA.
Citations
This is a case of tracheomalacia associated with esophageal atresia. An 11-month-old- male boy presented with a life-threatening apneic spell after correction of esophageal atresia (Gross type C). After complete exclusion of the other possible causes of the apneic spell, the presumptive diagnosis of tracheomalacia was made with fluoroscopy and 3-dimensional chest CT. The final diagnosis was made with rigid bronchoscopy under spontaneous respiration. The aortopexy was performed with intraoperative bronchoscopic examination. The postoperative period was unremarkably uneventful. The patient was discharged 9 days after the aortopexy and has remained well to date (5 months after the aortopexy).
Citations
When jaundice persists for more than 14 days postnatally, the early diagnosis of surgical jaundice is important for the prognosis in extrahepatic biliary atresia after draining procedure. The role of diagnostic laparoscopy to differenctiate medical causes of jaundice from biliary atresia is evaluated in this report. Four patients with prolonged jaundice have been included in this study. When the gallbladder was not visualized we proceeded to laparotomy. In patients with enlarged gallbladder visualized at laparoscopy, laparoscopic guided cholangiogram was performed, and laparoscopic liver biopsy was done for those who had a patent biliary tree. Two patients had small atretic gallbladder and underwent a Kasai hepato-portoenterostomy. One patients showed a patent gallbladder and common bile duct with atresia of the common hepatic and intrahepatic ducts, and they underwent a Kasai hepatic-portoenterostomy. One patient showed an enlarged gallbladder and laparoscopic-guided cholangiogram were normal. Laparoscopic liver biopsy was performed. There were no complications. Laparoscopy wth laparoscopic-guided cholangiogram may be a valuable method in accurate and earlier diagnosis in an infant with prolonged jaundice.
A survey on biliary atresia was made among 26 members of the Korean Association of Pediatric Surgeons. The members were required to complete a questionnaire and a case registration form for each patient during the twentyone-year period of 1980-2000. Three hundred and eighty patients were registered from 18 institutions. The average number of patients per surgeon was one to two every year. The male to female ratio was 1 : 1.3. The age of patients on diagnosis with biliary atresia was on average 65.4 ±36.2 days old. The national distribution was 32.8% in Seoul, 25.3% in Gyoungki-Do, 21.6% in Gyoungsang-Do, 9.27% in Choongchung-Do, etc. in order. The most common clinical presentation was jaundice (98.4%) and change of stool color (86.2%) was second. Two hundred eighty (74.7%) of 375 patients were operated by 80 days of age. Three hundred thirty six (91.9%) of 366 patients were operated on by the original Kasai procedure, and 305 (84.3%) of 362 patients were observed by bile-drainage postoperatively. The overall postoperative complication rate was 18.5% and the overall postoperative mortality rate was 6.8%. The associated anomalies were observed in 72 cases (22.5%). One hundred ninty five (64.7%) of 302 patients have been alive in follow-up and 49 (25.1%) have survived over 5 years without problem after operation. Ascending cholangitis, varices and ascites affected survival significantly, and the important long-term prognostic factor was the occurrence of complications.
Citations
 First
First Prev
Prev